NM_000548.5:c.4006-8C>T is an intronic variant at position -8 of exon 34 in TSC2, not predicted to alter splicing (SpliceAI delta = 0.00).1 This variant is present in gnomAD at appreciable population frequencies: v2.1 AF=0.2436% (538/220866 alleles, 5 homozygotes) and v4.1 AF=0.3818% (6017/1575890 alleles, 15 homozygotes), with the highest subpopulation frequency in the European (non-Finnish) group at 0.3946% (v2.1) and 0.4712% (v4.1). These frequencies far exceed the expected prevalence of tuberous sclerosis complex (estimated incidence ~1:6000 to 1:10000 live births), meeting BS1 (strong benign).2 Five homozygotes are observed in gnomAD v2.1 and 15 homozygotes in gnomAD v4.1. TSC is an autosomal dominant disorder with near-complete penetrance and significant morbidity; the presence of homozygotes in a general population database is incompatible with pathogenicity, meeting BS2 (strong benign).3 This variant has been classified as Benign by 14 clinical diagnostic laboratories and as Likely benign by 5 clinical laboratories in ClinVar (Variation ID 49281), meeting BP6 (supporting benign).4 SpliceAI predicts no splicing impact (max delta = 0.00), and the variant is an intronic substitution at a non-canonical splice position, meeting BP4 and BP7 (each supporting benign).5 No variant-specific functional studies, de novo observations, cosegregation data, or case-control studies were identified for this variant. The ClinVar criterion-lead submission (SCV000066333, Tuberous sclerosis database) cited PMID:17304050, but the full text of that publication does not mention NM_000548.5:c.4006-8C>T. Classification: Likely Benign. Benign evidence includes BS1 (strong), BS2 (strong), BP4 (supporting), BP6 (supporting), and BP7 (supporting). No pathogenic or likely pathogenic criteria are met. Under the ACMG/AMP 2015 combining criteria, ≥2 strong benign criteria → Likely Benign.6
TSC2
Final classification
Benign
TSC2 c.4006-8C>T · p.?
TSC2
NM_000548.5:c.4006-8C>T is an intronic variant at position -8 of exon 34 in TSC2, not predicted to alter splicing (SpliceAI delta = 0.00).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong, BS2 strong, BP4 supporting benign, BP6 supporting benign, BP7 supporting benign; combination = 2 strong benign + 3 supporting benign, which maps to Benign.
Classification rationale
BS1BS2BP4BP6BP7
Benign
TSC2 c.4006-8C>T
BS1 + BS2 + BP4 + BP6 + BP7
→
Benign
6
generic_acmg_combination_rules
Gene diagram
· NM_000548.5 · variants mapped to exon structure
TSC2
NM_000548.5
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 15 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
This variant is present in gnomAD at allele frequencies exceeding the expected prevalence of tuberous sclerosis complex. The NFE subpopulation AF is 0.3946% in v2.1 (365/92500 alleles, 4 homozygotes) and 0.4712% in v4.1 (5465/1159748 alleles, 13 homozygotes). TSC has an estimated incidence of ~1:6000 to 1:10000 live births, making the observed population frequency incompatible with a highly penetrant pathogenic variant. Under generic ACMG rules, BS1 applies when AF exceeds the expected disease frequency (>0.3% threshold used here).
gnomAD v2.1 NFE AF = 0.3946%exceeds BS1 >0.3% thresholdgnomAD v4.1 NFE AF = 0.4712%
✓
BS2
strong
Benign
This variant has been observed in a homozygous state in ostensibly healthy population controls — 5 homozygotes in gnomAD v2.1 and 15 homozygotes in gnomAD v4.1. Tuberous sclerosis complex is an autosomal dominant disorder with near-complete penetrance and significant morbidity. The presence of homozygotes in a general population database is incompatible with a pathogenic role, providing strong evidence for a benign classification.
5 homozygotes in gnomAD v2.1 (total: 538/220866 alleles)15 homozygotes in gnomAD v4.1 (total: 6017/1575890 alleles)TSC is a highly penetrant autosomal dominant disorder
✓
BP4
supporting
Benign
SpliceAI predicts no significant splicing impact for this intronic variant (max delta score = 0.00 across acceptor gain, acceptor loss, donor gain, and donor loss). Multiple in silico tools for splice prediction (SpliceAI delta = 0.00) agree that this variant does not alter normal splicing, supporting a benign interpretation.
SpliceAI max delta = 0.00 (no splice impact predicted)Variant is at intronic position -8SpliceAI predicts no cryptic splice site creation or canonical splice site disruption
✓
BP6
supporting
Benign
This variant has been classified as Benign by 14 clinical diagnostic laboratories and as Likely benign by 5 clinical diagnostic laboratories in ClinVar (Variation ID 49281). Multiple independent, reputable clinical testing laboratories have reached a consensus benign interpretation. The ClinVar review status is 'criteria provided, single submitter' for individual submissions, but the aggregate consensus across 19 clinical laboratories provides supporting evidence for a benign classification.
ClinVar Variation ID 49281Benign: 14 clinical laboratoriesLikely benign: 5 clinical laboratories
✓
BP7
supporting
Benign
c.4006-8C>T is an intronic variant located at position -8 of exon 34. SpliceAI predicts no impact on splicing (max delta = 0.00), indicating the variant does not affect the splice consensus sequence nor create a cryptic splice site. Under ACMG/AMP guidelines, intronic variants at non-conserved positions with no predicted splicing effect may be classified as BP7 (supporting benign).
Intronic variant at c.4006-8 (non-canonical splice position)SpliceAI delta = 0.00 confirms no predicted splicing alterationNo evolutionary conservation data suggest functional importance of this nucleotide position
Assessed · not applied
Pathogenic
PS2
No de novo occurrence of NM_000548.5:c.4006-8C>T with confirmed paternity has been identified in ClinVar criterion-lead submissions, published literature, or exploratory evidence recovery.
PS3
No functional studies specifically assaying NM_000548.5:c.4006-8C>T (e.g., minigene splicing reporter, RT-PCR, protein function) were identified in literature, ClinVar criterion-lead submissions, or exploratory evidence recovery.
PS4
No case-control studies comparing the prevalence of NM_000548.5:c.4006-8C>T in TSC-affected individuals versus population controls were identified.
PM1
c.4006-8 lies in intron 33, near exon 34.
PM2
This variant is present in gnomAD at appreciable frequencies: v2.1 overall AF=0.2436% (538/220866 alleles, 5 homozygotes) with NFE AF=0.3946%; v4.1 overall AF=0.3818% (6017/1575890 alleles, 15 homozygotes) with NFE AF=0.4712%.
PM6
No de novo occurrence of NM_000548.5:c.4006-8C>T without confirmed paternity/maternity was identified.
PP1
No cosegregation data are available for NM_000548.5:c.4006-8C>T in families with tuberous sclerosis complex.
PP3
In silico tools do not predict a deleterious effect for this variant.
PP4
No patient phenotype or family history data specific to NM_000548.5:c.4006-8C>T were available for review.
PP5
PP5 requires a reputable source (e.g., clinical diagnostic laboratory with recognized expertise) to report the variant as pathogenic.
Benign
BA1
BA1 requires allele frequency >1% in a general population database.
BS3
No variant-specific functional studies demonstrating a benign effect (e.g., normal splicing by RT-PCR, retained protein function) were identified for NM_000548.5:c.4006-8C>T.
BS4
No data are available regarding non-segregation of NM_000548.5:c.4006-8C>T with tuberous sclerosis complex in affected families.
BP2
No observation of NM_000548.5:c.4006-8C>T in trans with a known pathogenic TSC2 variant has been identified.
BP5
No data are available indicating that NM_000548.5:c.4006-8C>T has been observed in a patient with an alternate molecular basis for tuberous sclerosis complex.
N/A · 6
PVS1 · PS1 · PM5 · PP2 · BP1 · BP3
Research & evidence
Population frequency · supports benign
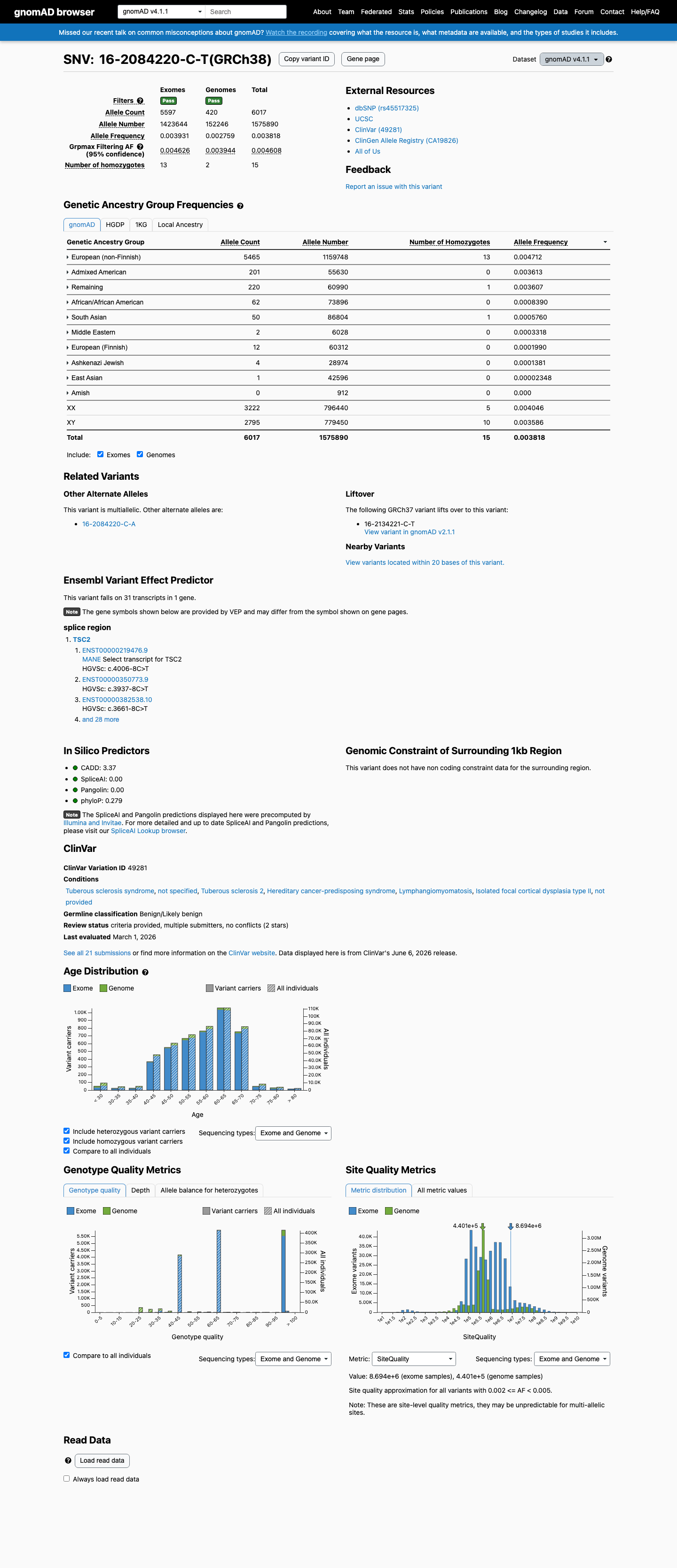
gnomAD v4.1
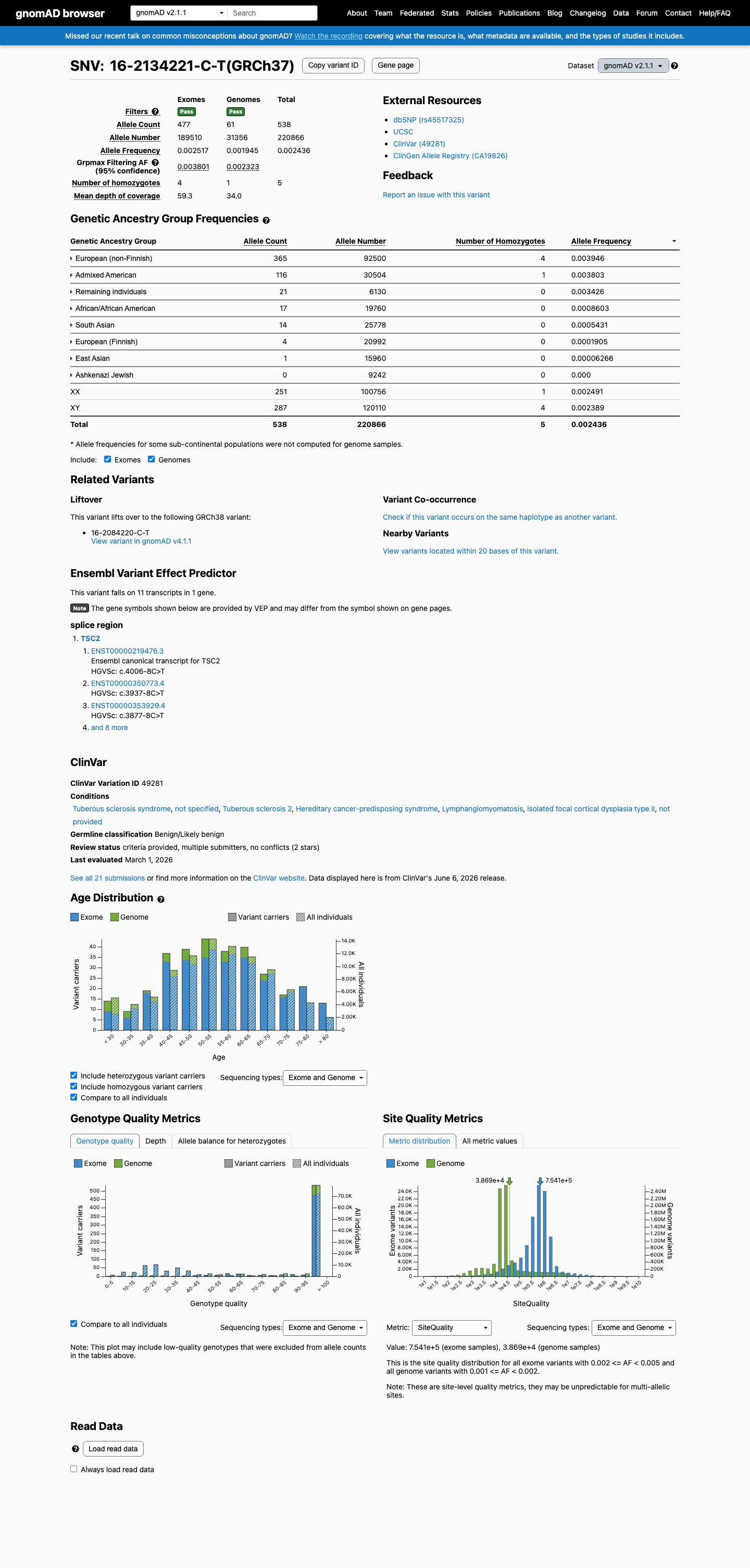
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00381816; MAF= 0.38182%, 6017/1575890 alleles, homozygotes = 15) and has highest observed frequency in the European (non-Finnish) population (AF= 0.00471223; MAF= 0.47122%, 5465/1159748 alleles, homozygotes = 13); grpmax FAF= 0.0046076.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00243587; MAF= 0.24359%, 538/220866 alleles, homozygotes = 5) and has highest observed frequency in the European (non-Finnish) population (AF= 0.00394595; MAF= 0.39459%, 365/92500 alleles, homozygotes = 4); grpmax FAF= 0.00380144.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.38%
· 6017 / 1,575,890
15 hom · FAF 0.46%
15 hom · FAF 0.46%
European (non-Finnish) 5465 / 1,159,748 |
0.47% 13 hom |
Admixed American 201 / 55,630 |
0.36% |
Remaining individuals 220 / 60,990 |
0.36% 1 hom |
African/African American 62 / 73,896 |
0.084% |
South Asian 50 / 86,804 |
0.058% 1 hom |
Middle Eastern 2 / 6,028 |
0.033% |
European (Finnish) 12 / 60,312 |
0.02% |
Ashkenazi Jewish 4 / 28,974 |
0.014% |
East Asian 1 / 42,596 |
0.0023% |
+ 1 not observed (Amish)
gnomAD v2.1
0.24%
· 538 / 220,866
5 hom · FAF 0.38%
5 hom · FAF 0.38%
European (non-Finnish) 365 / 92,500 |
0.39% 4 hom |
Admixed American 116 / 30,504 |
0.38% 1 hom |
Remaining individuals 21 / 6,130 |
0.34% |
African/African American 17 / 19,760 |
0.086% |
South Asian 14 / 25,778 |
0.054% |
European (Finnish) 4 / 20,992 |
0.019% |
East Asian 1 / 15,960 |
0.0063% |
+ 1 not observed (Ashkenazi Jewish)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
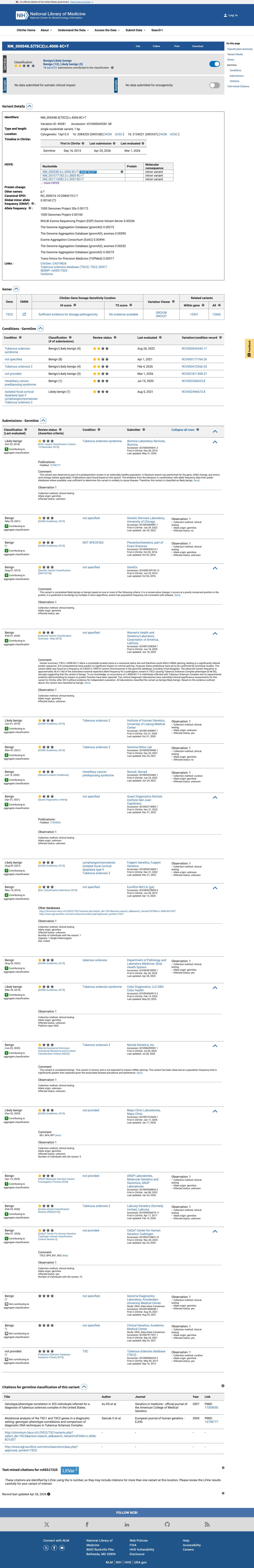
ClinVar
This variant has been reported in ClinVar as Benign (14 clinical laboratories) and as Likely benign (5 clinical laboratories). (ClinVarID = 49281)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV104573703, n = 2 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
15798777 ↗
Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex.
CLINVAR
17304050 ↗
Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
23519317 ↗
Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions.
CLINVAR
23788249 ↗
ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.
CLINVAR
25356965 ↗
ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR