NM_004304.4:c.4033G>A (p.Gly1345Arg) is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (AF 1.86 × 10⁻⁶, 3/1,613,830 alleles), meeting PM2 at supporting strength.1 The variant is a missense substitution not located in a statistically significant mutational hotspot; PM1 is not met. In silico predictions are conflicting: REVEL score 0.786 supports a deleterious effect, but BayesDel score 0.144 is in the benign range and SpliceAI max delta 0.20 is borderline. PP3 is not met, and BP4 is not met.2 This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory, SCV002134937); no reputable source classifies it as pathogenic or benign. PP5 and BP6 are not met.3 No variant-specific functional studies (PS3/BS3), segregation data (PP1/BS4), de novo observations (PS2/PM6), or case-control data (PS4) were identified. PVS1 is not applicable as this is a missense variant outside canonical null-variant categories.4 With only PM2_Supporting met, the variant does not reach a Likely Pathogenic threshold (requiring at least 2 Supporting criteria or 1 Moderate + 1 Supporting under generic ACMG/AMP rules) nor a Likely Benign threshold. The variant remains a Variant of Uncertain Significance.5
ALK
Final classification
VUS
ALK c.4033G>A · p.Gly1345Arg
ALK
NM_004304.4:c.4033G>A (p.Gly1345Arg) is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (AF 1.86 × 10⁻⁶, 3/1,613,830 alleles), meeting PM2 at supporting strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting; combination = 1 supporting, which maps to VUS.
Classification rationale
PM2
VUS
ALK c.4033G>A
PM2
→
VUS
2
revelbayesdelspliceai ↗
4
pvs1_generic_framework ↗oncokb ↗pvs1_variant_assessment
5
generic_acmg_combination_rules
Gene diagram
· NM_004304.4 · variants mapped to exon structure
ALK
NM_004304.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 21 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
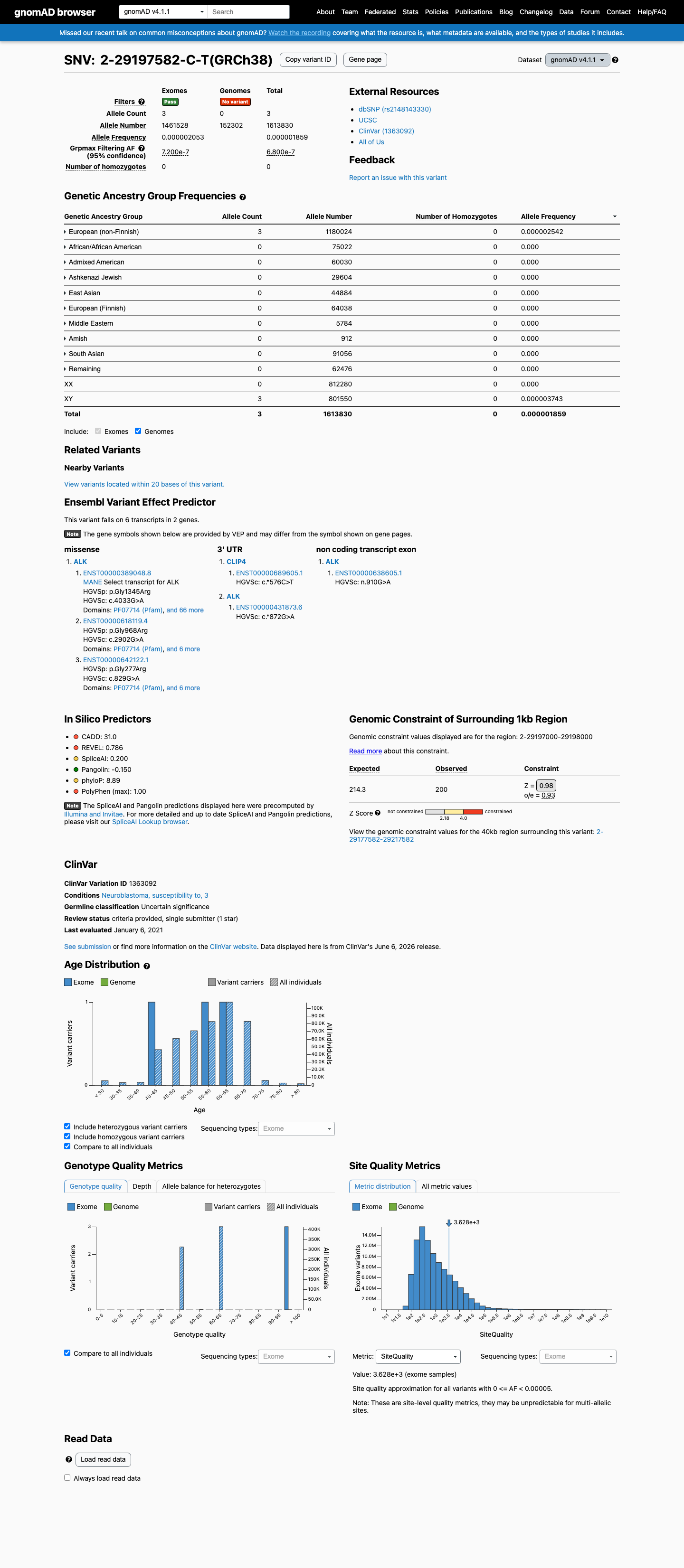
NM_004304.4:c.4033G>A is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (overall AF = 1.86 × 10⁻⁶, 3/1,613,830 alleles; highest subpopulation frequency = 2.54 × 10⁻⁶ in European non-Finnish; grpmax FAF = 6.8 × 10⁻⁷). This frequency is well below the non-VCEP PM2 supporting threshold of <0.1%.
gnomAD v2.1: absent. gnomAD v4.1: AF 1.86e-06 (0.00019%)3 alleles total in NFE subpopulation0 homozygotes
Assessed · not applied
Pathogenic
PS1
No evidence was identified for an established pathogenic variant with the same amino acid change (p.Gly1345Arg) resulting from a different nucleotide substitution.
PS2
No de novo observation with confirmed maternity and paternity was identified for this variant in any source.
PS3
No variant-specific well-established in vitro or in vivo functional studies were identified.
PS4
No case-control studies or cohort data comparing variant prevalence in affected versus unaffected individuals were identified.
PM1
The variant is not located in a statistically significant mutational hotspot.
PM6
No assumed de novo observations (without confirmed maternity/paternity) were identified for this variant in any source.
PP1
No cosegregation data in multiple affected family members were identified for this variant.
PP2
HCI prior probability is not available for ALK (gene not supported).
PP3
In silico predictions are conflicting and do not provide multiple concordant lines of computational evidence for a deleterious effect.
PP4
No patient phenotype or family history data are available to assess whether the phenotype is highly specific for a disease with a single genetic etiology.
PP5
No reputable source classifies this variant as pathogenic.
Benign
BA1
The variant is absent from gnomAD v2.1 and present at AF = 1.86 × 10⁻⁶ in gnomAD v4.1, far below the BA1 threshold of >1% (and the more conservative >5%).
BS1
The variant frequency in gnomAD v4.1 (AF = 1.86 × 10⁻⁶, 0.00019%) is far below the non-VCEP BS1 threshold of >0.3%.
BS2
Observation of the variant at very low frequency in population databases (3 heterozygous alleles in gnomAD v4.1) does not meet BS2, which requires confirmed observation in a healthy adult individual for a disorder expected to be fully penetrant at an early age.
BS3
No well-established in vitro or in vivo functional studies demonstrating no damaging effect on protein function or splicing were identified for this variant.
BS4
No segregation data in affected family members are available to assess lack of cosegregation with disease.
BP1
ALK-related neuroblastic tumor susceptibility is not a disorder where only truncating variants cause disease.
BP2
No data are available regarding observation of this variant in trans with a pathogenic variant for a fully penetrant dominant disorder.
BP4
Multiple lines of computational evidence do not concordantly suggest no impact on the gene product.
BP5
No data are available regarding observation of this variant in a case with an alternate molecular basis for disease.
BP6
No reputable source classifies this variant as benign.
N/A · 4
PVS1 · PM5 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.85893e-06; MAF= 0.00019%, 3/1613830 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 2.54232e-06; MAF= 0.00025%, 3/1180024 alleles, homozygotes = 0); grpmax FAF= 6.8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00019%
· 3 / 1,613,830
0 hom · FAF 6.8e-05%
0 hom · FAF 6.8e-05%
European (non-Finnish) 3 / 1,180,024 |
0.00025% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
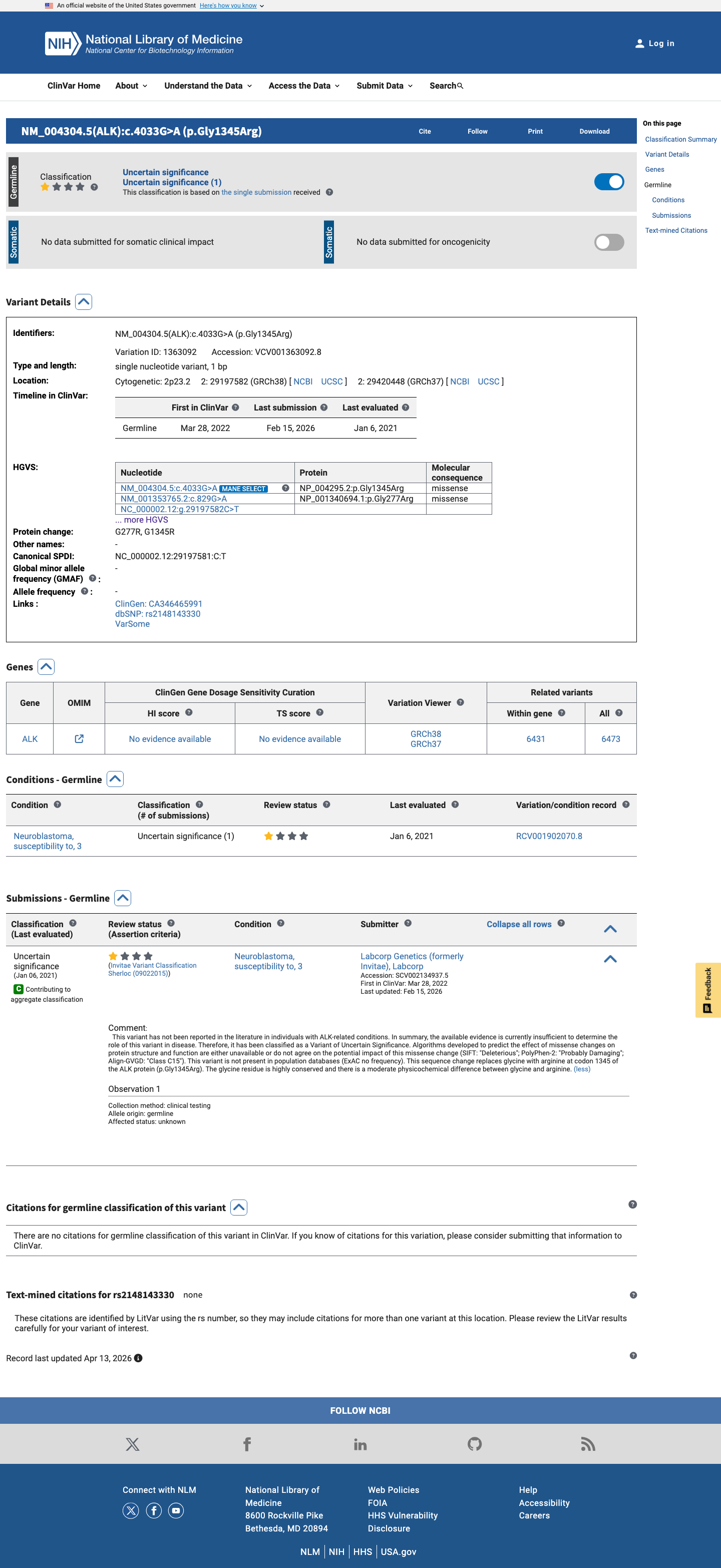
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory). (ClinVarID = 1363092)
In silico
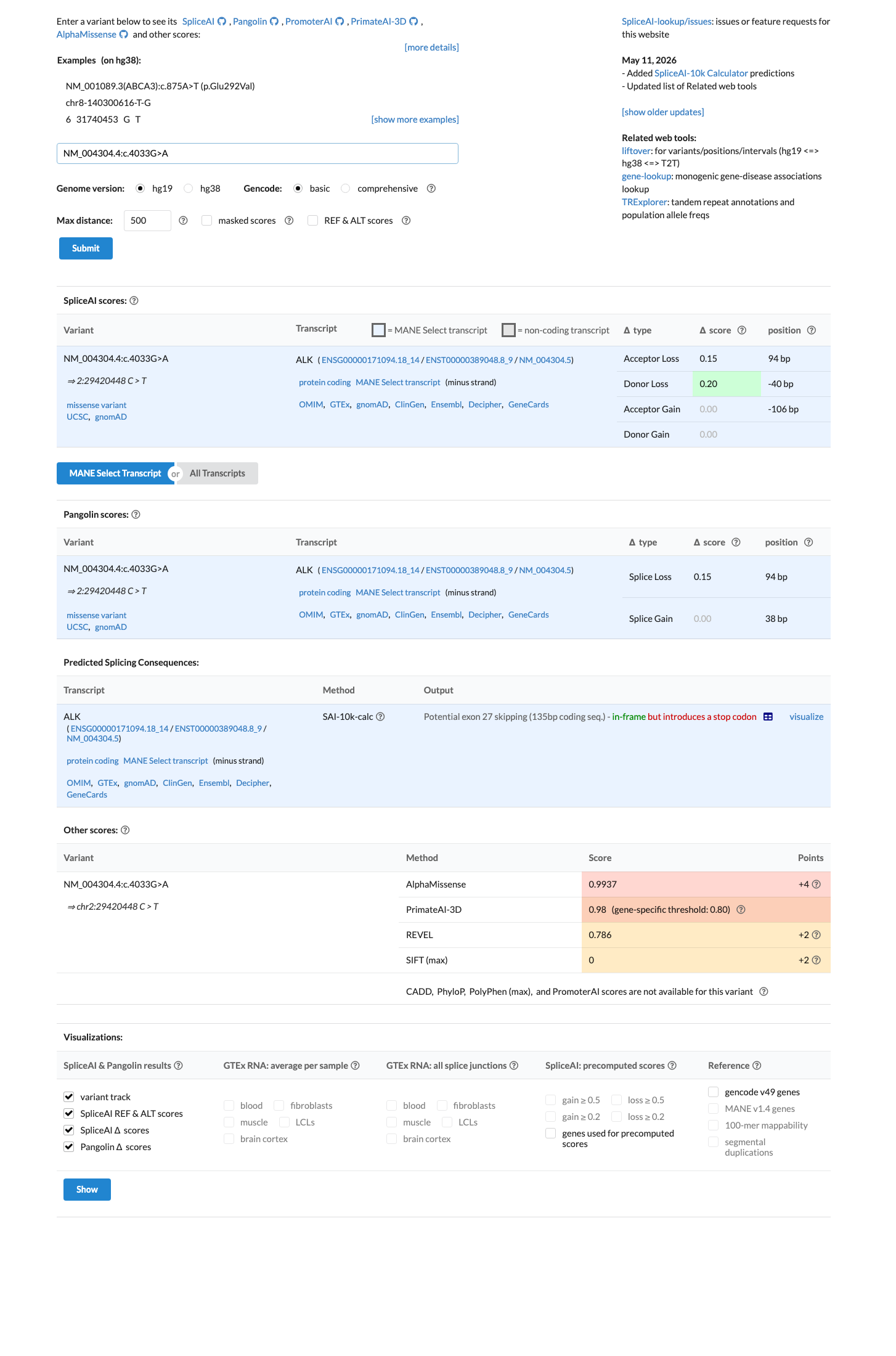
SpliceAI predicts possible splice impact for this variant (max delta score = 0.20). REVEL score = 0.786. BayesDel score = 0.144187.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. ALK, a receptor tyrosine kinase, is recurrently altered by chromosomal rearrangements in various cancer types including anaplastic large cell lymphoma

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR