NM_203407.3:c.616G>A (p.Ala206Thr) in EZHIP is absent from ClinVar.1 The variant is extremely rare in population databases, absent from gnomAD v2.1 (0/162,702 alleles) and present as a singleton in gnomAD v4.1 (1/563,095 alleles, AF=0.000178%), meeting PM2 at supporting strength.2 p.Ala206Thr lies within the PRC2-binding domain of EZHIP (residues 200–391), a well-characterized functional domain where pathogenic missense variants cluster, meeting PM1 at supporting strength. SpliceAI predicts no splicing impact (max delta=0.01) and BayesDel yields a benign-leaning score of −0.673766, meeting BP4 at supporting benign strength.3 No functional studies, de novo observations, co-segregation data, or case-control evidence are available for this variant. No reputable source classification exists.4 With two supporting pathogenic criteria (PM1_supporting, PM2_supporting) and one supporting benign criterion (BP4_supporting_benign), the evidence is insufficient to classify the variant as pathogenic or benign. The variant is classified as a Variant of Uncertain Significance (VUS).5
EZHIP
Final classification
VUS
EZHIP c.616G>A · p.Ala206Thr
EZHIP
NM_203407.3:c.616G>A (p.Ala206Thr) in EZHIP is absent from ClinVar.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 supporting, PM2 supporting, BP4 supporting benign; combination = 2 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM1PM2
BP4
VUS
EZHIP c.616G>A
PM1 + PM2 + BP4
→
VUS
3
spliceai ↗bayesdel
5
generic_acmg_combination_rules
Gene diagram
· NM_203407.3 · variants mapped to exon structure
EZHIP
NM_203407.3
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 20 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
supporting
review
Pathogenic
p.Ala206Thr is located within the PRC2-binding domain of EZHIP (residues 200–391), a well-characterized functional domain where pathogenic missense variants cluster in patients with overgrowth syndrome. The domain is defined by structural studies (PMID:32325000) and is the site of multiple de novo missense variants reported in PMID:31548708. Position 206 is at the N-terminal boundary of this domain; applied at supporting rather than moderate strength given the position is at the domain edge and no residue-specific constraint data are available.
Residue 206 falls within the PRC2-binding domain (residues 200–391)a critical functional domainMultiple pathogenic/de novo missense variants reported in this domain (PMID:31548708
✓
PM2
supporting
Pathogenic
NM_203407.3:c.616G>A is extremely rare in large population databases. In gnomAD v2.1 it is absent (0/162,702 alleles). In gnomAD v4.1 it is observed as a singleton (1/563,095 alleles, AF=1.78e-06, no homozygotes). Both are well below the PM2 threshold of 0.1% (non-VCEP). Absent from gnomAD-Canada v1.0.
gnomAD v2.1: 0/162702 alleles (0%)gnomAD v4.1: 1/563
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no significant impact. SpliceAI predicts no splicing alteration (max delta=0.01). BayesDel score is −0.673766, leaning toward a benign interpretation. Although REVEL is unavailable, two independent in silico tools converge on a neutral/benign prediction. Applied at supporting benign strength due to incomplete in silico coverage (REVEL missing).
SpliceAI max delta: 0.01 (no predicted splicing impact)BayesDel score: −0.673766 (benign-leaning prediction)
Assessed · not applied
Pathogenic
PS1
PS1 applies when the same amino acid change has been previously established as pathogenic.
PS2
No de novo observation of NM_203407.3:c.616G>A with confirmed parentage has been reported.
PS3
No well-established in vitro or in vivo functional studies have been performed on p.Ala206Thr.
PS4
No case-control study or enrichment analysis demonstrates a statistically significant excess of this variant in affected individuals versus controls.
PM5
No pathogenic missense variant at the same residue (p.Ala206) was identified in ClinVar.
PM6
No de novo observation of c.616G>A without confirmed parentage has been reported.
PP1
No co-segregation data available.
PP2
PP2 requires a missense variant in a gene with a low rate of benign missense variation (high missense Z-score) where missense variants are a common mechanism of disease.
PP3
Multiple in silico tools do not support a deleterious effect.
PP4
No patient phenotype or family history data are available for the individual carrying this variant.
PP5
No reputable source (clinical laboratory, expert panel) has classified this variant as pathogenic.
Benign
BA1
BA1 requires a minor allele frequency >1% in population databases.
BS1
BS1 requires a minor allele frequency >0.3% (non-VCEP cutoff).
BS2
BS2 requires observation of the variant in healthy adults with full penetrance expected at an early age.
BS3
No well-established functional studies exist demonstrating no damaging effect of p.Ala206Thr on protein function or splicing.
BS4
No data demonstrating lack of segregation of this variant with disease in affected families.
BP1
BP1 applies to a missense variant in a gene where only truncating variants cause disease.
BP2
No observation of this variant in trans with a known pathogenic EZHIP variant.
BP5
No evidence that this variant was found in a case with an alternate molecular basis for disease.
BP6
No reputable source has classified this variant as benign.
N/A · 3
PVS1 · BP3 · BP7
Research & evidence
Population frequency
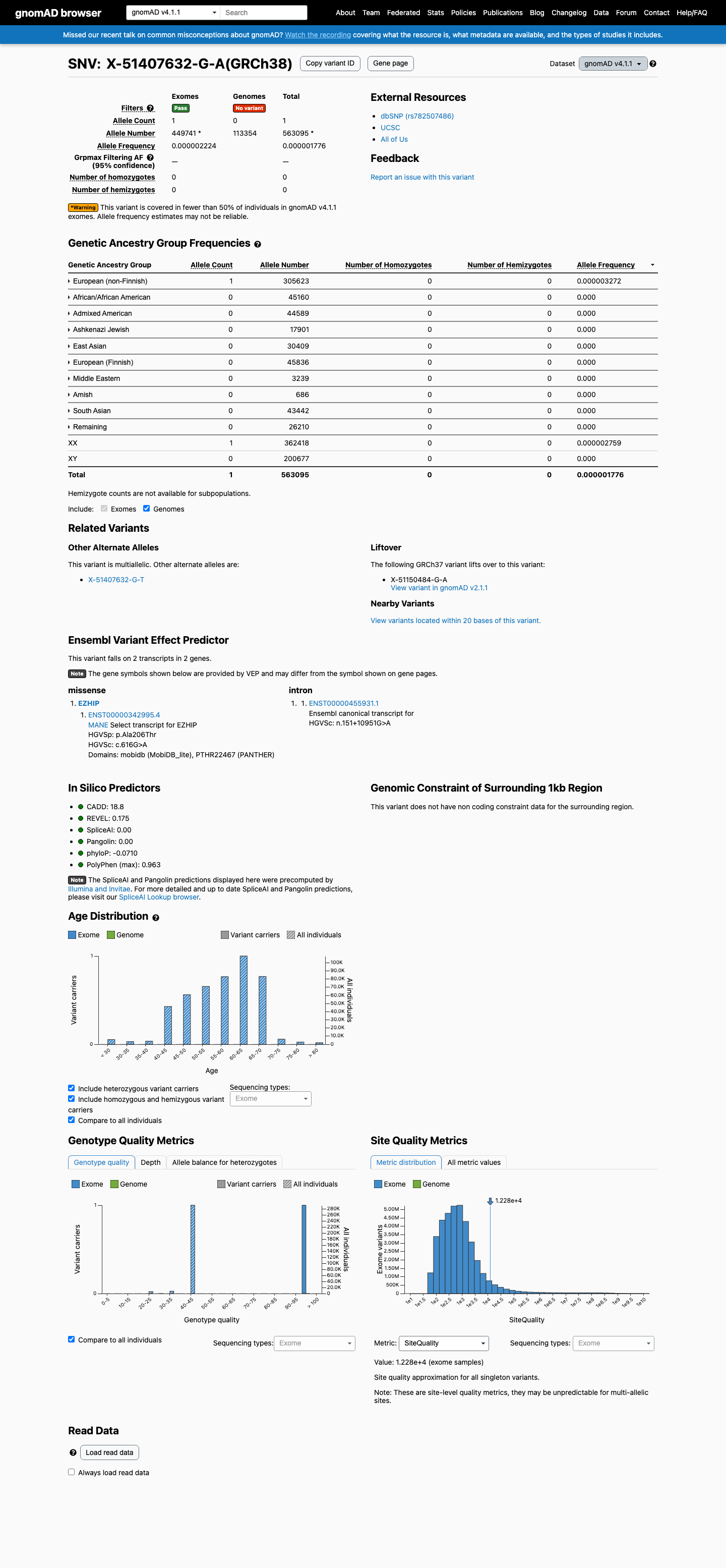
gnomAD v4.1
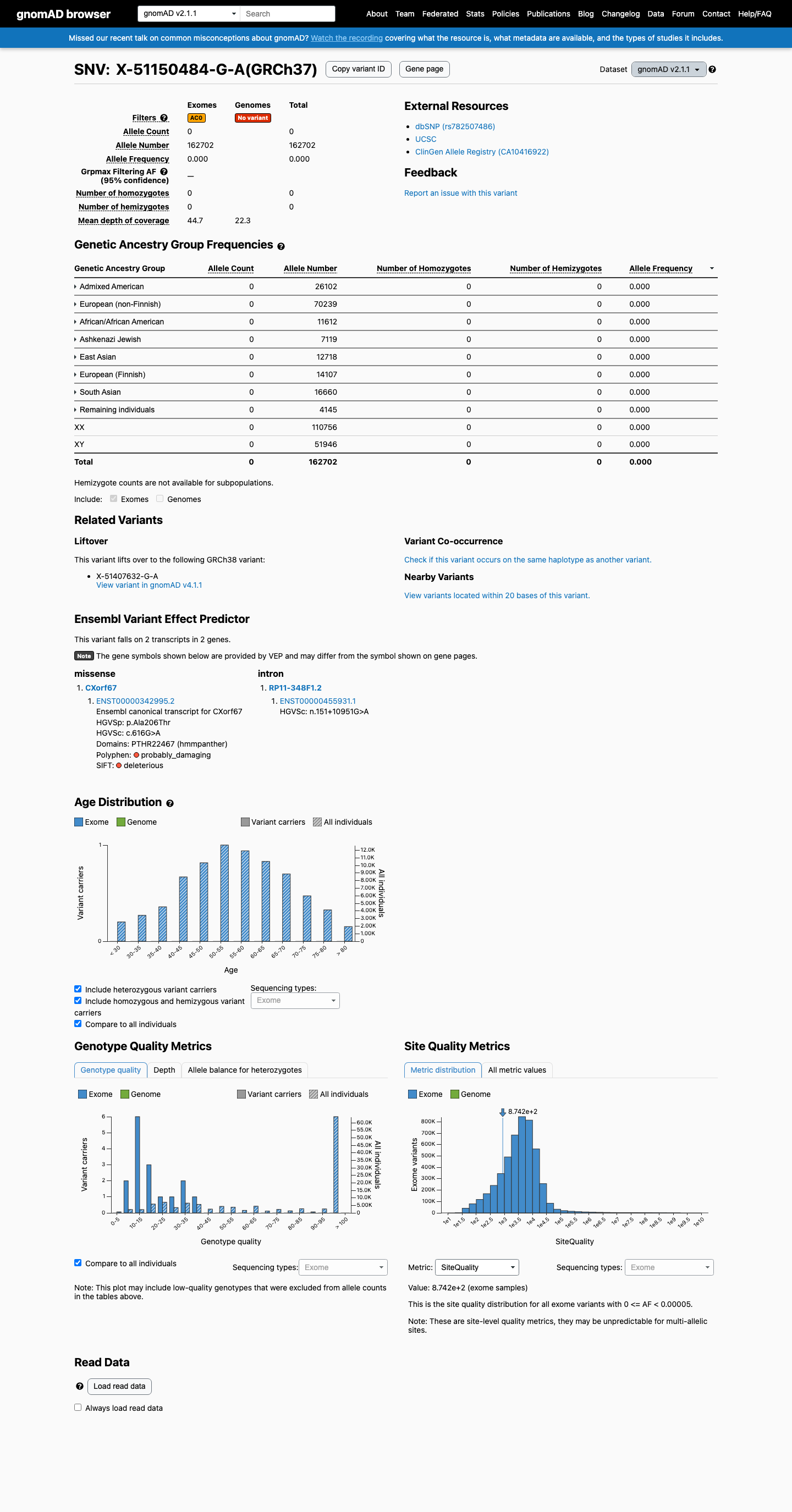
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.7759e-06; MAF= 0.00018%, 1/563095 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.27201e-06; MAF= 0.00033%, 1/305623 alleles, homozygotes = 0).
v2.1
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/162702 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/11612 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00018%
· 1 / 563,095
0 hom
0 hom
European (non-Finnish) 1 / 305,623 |
0.00033% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / 162,702
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
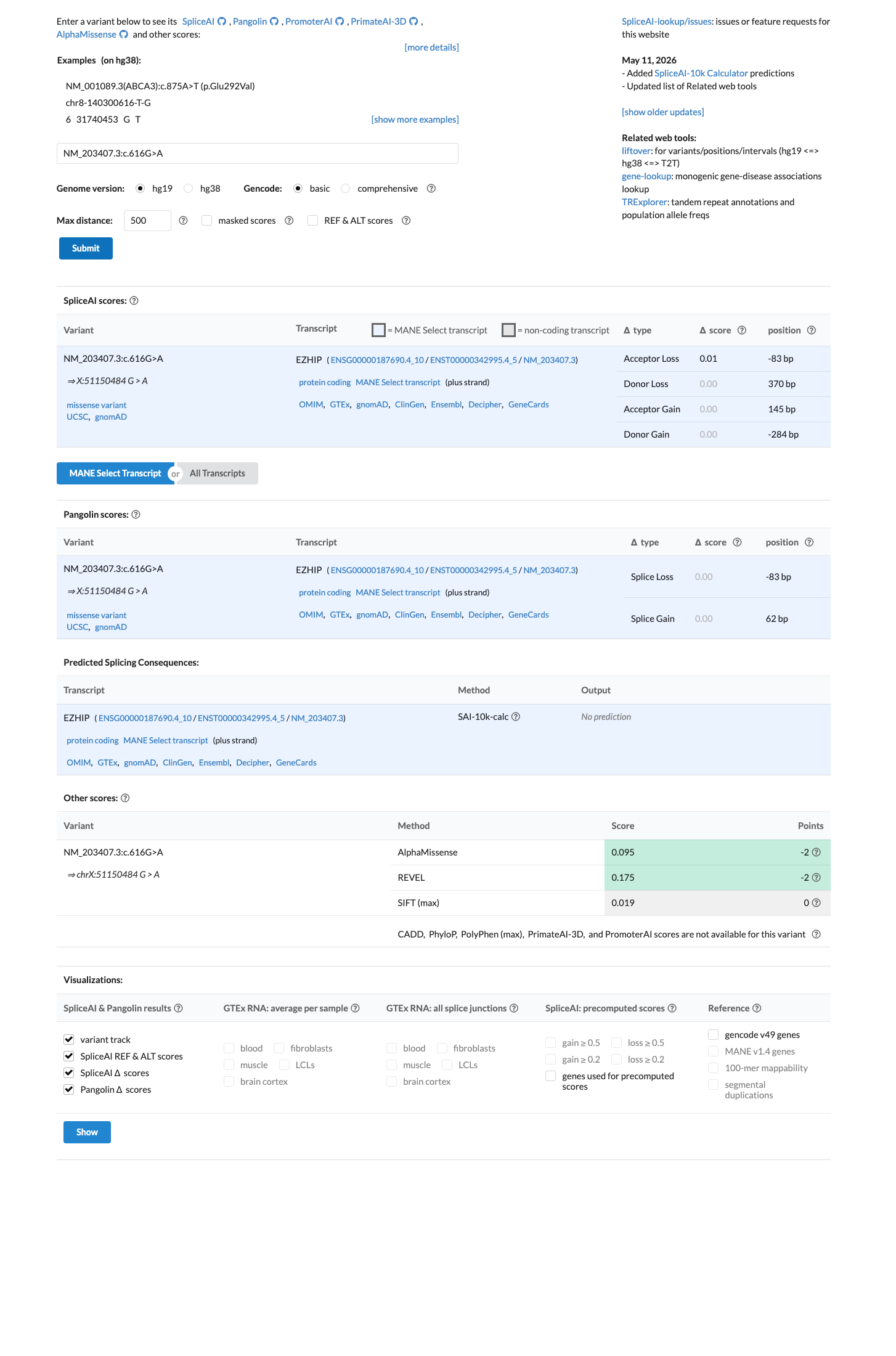
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). BayesDel score = -0.673766.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. EZHIP, a polycomb binding protein, is recurrently altered by rearrangement and mutation in endometrial stromal sarcomas and posterior fossa ependymoma

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
PMID 31548708
Found
Residue 206 falls within the PRC2-binding domain (residues 200–391) a critical functional domain Multiple pathogenic/de novo missense variants reported in this domain (PMID:31548708 PMID:32325000)
Applied to
→PM1 supports · met
PMID 32325000
Found
Residue 206 falls within the PRC2-binding domain (residues 200–391) a critical functional domain Multiple pathogenic/de novo missense variants reported in this domain (PMID:31548708 PMID:32325000)
Applied to
→PM1 supports · met
Sources & reference links