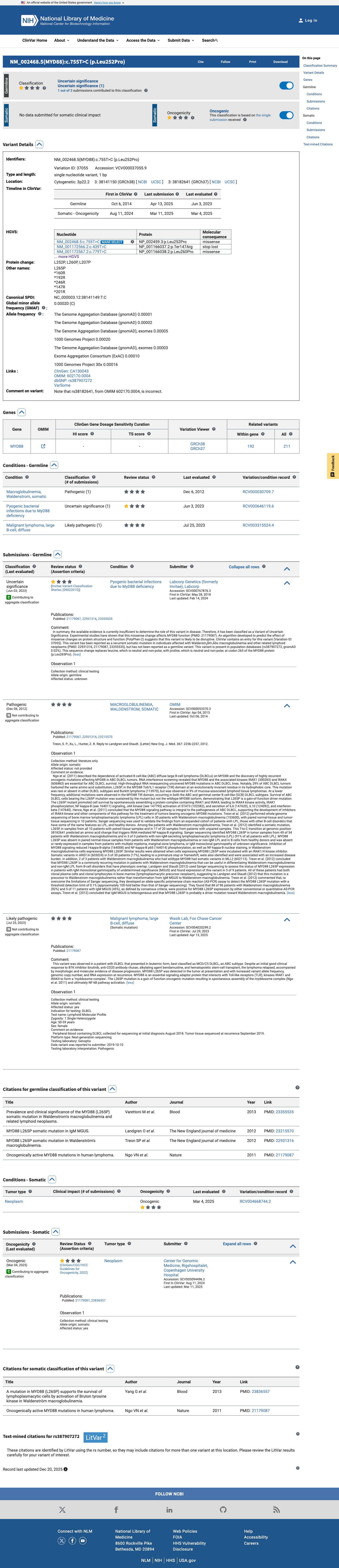
MYD88 c.818T>C (p.Leu273Pro), corresponding to the canonical L265P mutation, is a well-characterized gain-of-function missense variant in the TIR domain BB-loop (PM1_moderate).1 Multiple independent functional studies demonstrate that this variant constitutively activates NF-kappaB and JAK/STAT signaling, confers cytokine-independent survival, and that mutant-specific knockdown or pharmacologic inhibition is selectively toxic to MYD88-mutant cells (PS3_strong).2 The variant is highly enriched in affected individuals, detected in approximately 90% of Waldenstrom macroglobulinemia cases and recurrent in ABC DLBCL and IgM-MGUS, compared to an extremely low population frequency of 0.0026-0.0052% in gnomAD with zero homozygotes (PS4_strong).3 The variant is absent or extremely rare in population databases, with gnomAD v2.1 AF=5.17e-05 and v4.1 AF=2.60e-05, both well below the 0.1% threshold (PM2_supporting).4 In silico predictors support a deleterious effect: REVEL score of 0.735 exceeds the pathogenicity threshold of 0.5, though BayesDel (0.13148) is borderline and SpliceAI predicts no splicing impact (PP3_supporting).5 All benign criteria were assessed and none were met. BS3 is specifically contradicted by well-established functional evidence of a gain-of-function damaging effect. BA1 and BS1 are not met as population frequencies remain well below benign thresholds.6 Applying generic ACMG/AMP 2015 combination rules: 2 Strong (PS3, PS4) + 1 Moderate (PM1) + 2 Supporting (PM2, PP3) meets the threshold for Pathogenic (>=2 Strong).7 CAVEAT: The evidence supporting PS3_strong and PS4_strong derives predominantly from somatic tumor studies. The variant is almost exclusively observed as a somatic mutation in hematologic malignancies. The germline ACMG/AMP framework is being applied to a variant with a somatic disease mechanism. Classification should be interpreted with this context, and human review is recommended to confirm the appropriateness of this germline-classification for the clinical indication.
MYD88
Final classification
Pathogenic
MYD88 c.818T>C · p.Leu273Pro
MYD88
MYD88 c.818T>C (p.Leu273Pro), corresponding to the canonical L265P mutation, is a well-characterized gain-of-function missense variant in the TIR domain BB-loop (PM1_moderate).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PS4 strong, PM1 moderate, PM2 supporting, PP3 supporting; combination = 2 strong + 1 moderate + 2 supporting, which maps to Pathogenic.
Classification rationale
PS3PS4PM1PM2PP3
Pathogenic
MYD88 c.818T>C
PS3 + PS4 + PM1 + PM2 + PP3
→
Pathogenic
5
revelbayesdelspliceai ↗
7
generic_acmg_combination_rules
Gene diagram
· NM_001172567.1 · variants mapped to exon structure
MYD88
NM_001172567.1
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 15 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Well-established functional studies from multiple independent groups demonstrate that MYD88 L273P (canonical L265P) constitutively activates NF-kappaB and JAK/STAT signaling, confers IL-6 and IL-10 autocrine survival, and that knockdown or inhibition of the mutant protein is selectively toxic to MYD88-mutant lymphoma cells. This gain-of-function mechanism is consistent with the known disease mechanism in Waldenstrom macroglobulinemia and ABC DLBCL.
Ngo et al. 2011 (PMID:21179087): MYD88 L265P identified in ABC DLBCLconstitutively activates NF-kappaB and JAK/STATknockdown kills mutant cells.
✓
PS4
strong
review
Pathogenic
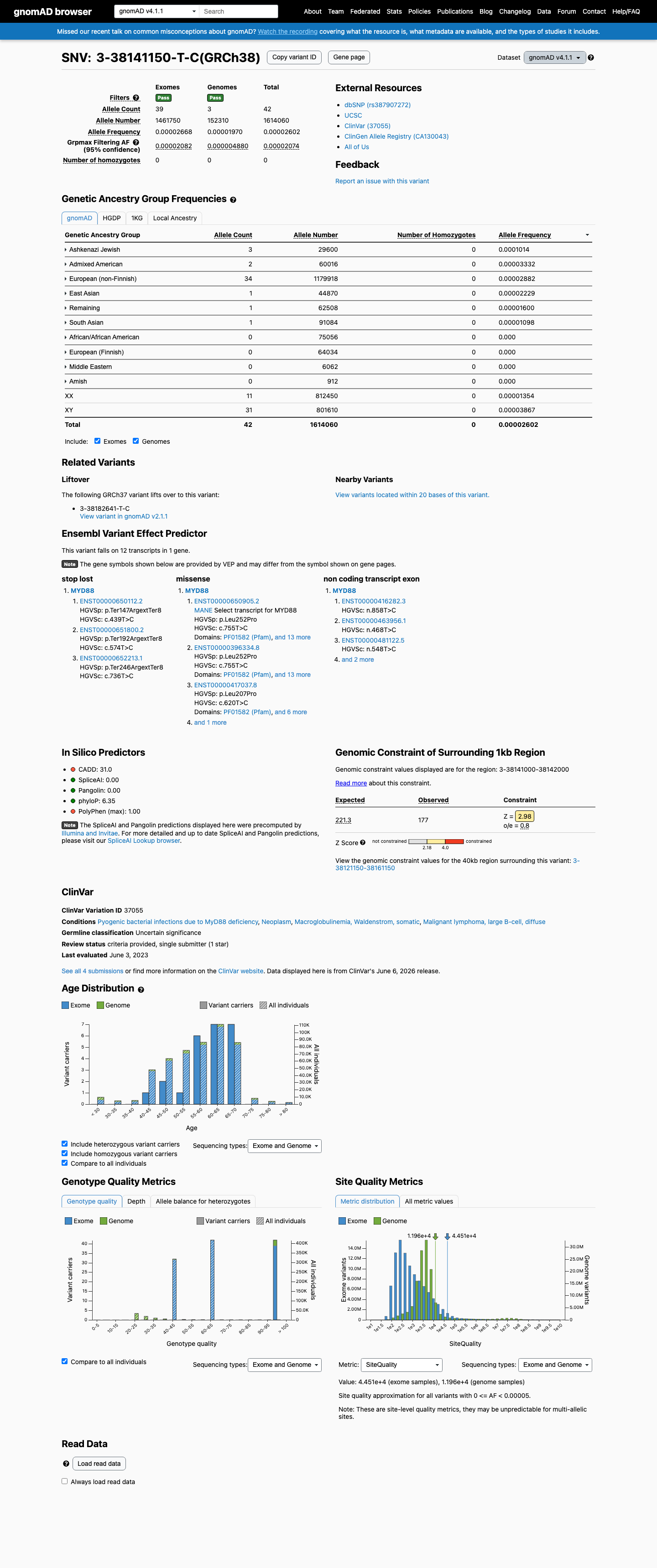
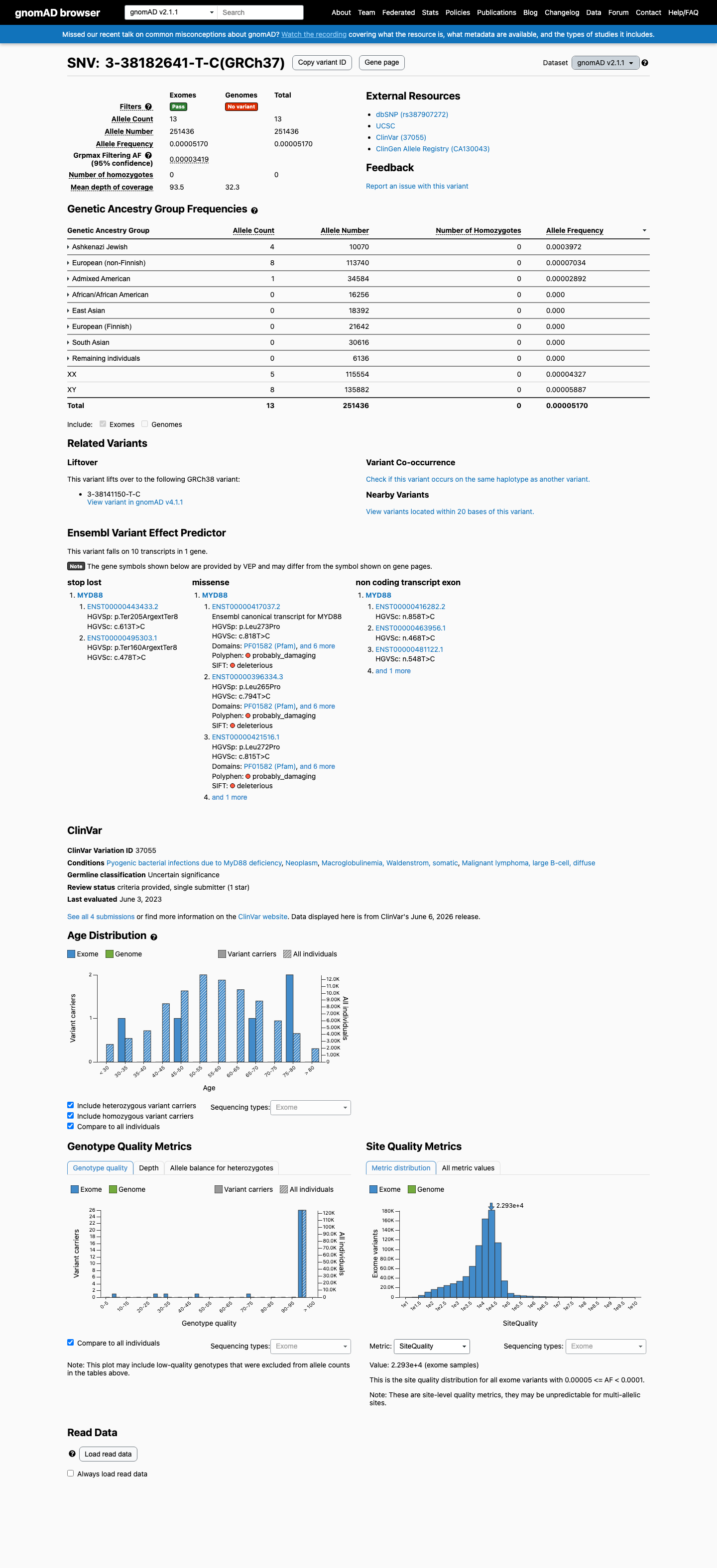
The variant is highly enriched in affected individuals compared to general population controls. MYD88 L273P (L265P) is detected in approximately 90% of Waldenstrom macroglobulinemia cases and is recurrent in ABC DLBCL and IgM-MGUS. In contrast, it is extremely rare in gnomAD (v2.1 AF=5.17e-05, 13/251,436 alleles; v4.1 AF=2.60e-05, 42/1,614,060 alleles; zero homozygotes). COSMIC reports 2,493 independent tumor samples with this variant. The odds ratio substantially exceeds the threshold for PS4_strong.
PMID:22931316: L265P in >90% of WM by whole-genome sequencing of 30 patients.PMID:23355535: L265P in 96% of WM54% of IgM-MGUS by allele-specific PCR.
✓
PM1
moderate
Pathogenic
The variant affects codon 273 within the TIR domain BB-loop, a critical functional domain essential for MYD88 signal transduction and a well-established mutational hotspot. COSMIC reports 2,493 independent samples with mutations in this domain.
Residue L273 is located in the TIR domain BB-loopa critical region for MYD88-mediated signaling.COSMIC: 2
✓
PM2
supporting
Pathogenic
The variant is extremely rare in population databases. gnomAD v2.1 allele frequency is 5.17e-05 (0.00517%, 13/251,436 alleles), and v4.1 allele frequency is 2.60e-05 (0.00260%, 42/1,614,060 alleles), both well below the 0.1% PM2 threshold. Zero homozygotes observed. gnomAD-Canada shows no data.
gnomAD v2.1: AF=5.17e-05 (13/251436 alleles0 homozygotes).
✓
PP3
supporting
Pathogenic
Multiple lines of in silico computational evidence support a deleterious effect. REVEL score is 0.735 (above the 0.5 threshold for pathogenicity). BayesDel score is 0.13148 (borderline/low). SpliceAI predicts no splicing impact (max delta = 0.0). The REVEL score provides supporting evidence for pathogenicity, though BayesDel is not strongly corroborative.
REVEL: 0.735 (deleterious>0.5 threshold).BayesDel: 0.13148 (borderline
Assessed · not applied
Pathogenic
PS2
No confirmed de novo germline occurrence (with both maternity and paternity confirmed) has been reported for this variant.
PM6
No evidence of assumed de novo occurrence (without confirmation of paternity and maternity).
PP1
No cosegregation data available.
PP2
Insufficient data available on MYD88 missense constraint metrics (e.g., gnomAD missense Z-score, OE ratio) to evaluate whether the gene has a low rate of benign missense variation.
PP4
No patient-specific phenotype or family history was provided for this case.
PP5
The only ClinVar submission with assertion criteria provided (Labcorp Genetics, SCV000767876) classifies this variant as Uncertain Significance, not Pathogenic.
Benign
BA1
Allele frequency is well below the 1% threshold for BA1.
BS1
Allele frequency is below the 0.3% threshold for BS1.
BS2
Although the variant is observed at very low frequency in gnomAD (presumed healthy population controls: 13-42 heterozygous carriers), this observation is at an allele frequency of 0.0026-0.0052%, which is far below the threshold for BS2.
BS3
Well-established functional studies demonstrate that MYD88 L273P (L265P) has a gain-of-function damaging effect (constitutive NF-kappaB activation, JAK/STAT signaling, cytokine secretion), not a benign effect.
BP1
MYD88-related disease (Waldenstrom macroglobulinemia, DLBCL) is driven by gain-of-function missense variants, not truncating loss-of-function variants.
BP2
No evidence of this variant being observed in trans with a pathogenic variant (for a dominant disorder) or in cis with a pathogenic variant (any inheritance pattern).
BP4
Multiple lines of in silico computational evidence do NOT support a benign interpretation.
BP5
No alternative molecular basis for disease has been identified in cases harboring this variant.
BP6
No reputable source classifies this variant as benign.
N/A · 8
PVS1 · PS1 · PM3 · PM4 · PM5 · BS4 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.60213e-05; MAF= 0.00260%, 42/1614060 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.000101351; MAF= 0.01014%, 3/29600 alleles, homozygotes = 0); grpmax FAF= 2.074e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 5.1703e-05; MAF= 0.00517%, 13/251436 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.000397219; MAF= 0.03972%, 4/10070 alleles, homozygotes = 0); grpmax FAF= 3.419e-05.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0026%
· 42 / 1,614,060
0 hom · FAF 0.0021%
0 hom · FAF 0.0021%
Ashkenazi Jewish 3 / 29,600 |
0.01% |
Admixed American 2 / 60,016 |
0.0033% |
European (non-Finnish) 34 / 1,179,918 |
0.0029% |
East Asian 1 / 44,870 |
0.0022% |
Remaining individuals 1 / 62,508 |
0.0016% |
South Asian 1 / 91,084 |
0.0011% |
+ 4 not observed (European (Finnish), Amish, Middle Eastern, African/African American)
gnomAD v2.1
0.0052%
· 13 / 251,436
0 hom · FAF 0.0034%
0 hom · FAF 0.0034%
Ashkenazi Jewish 4 / 10,070 |
0.04% |
European (non-Finnish) 8 / 113,740 |
0.007% |
Admixed American 1 / 34,584 |
0.0029% |
+ 5 not observed (African/African American, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.735. BayesDel score = 0.13148.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MYD88, an adaptor protein, is frequently altered in hematologic malignancies including Waldenström's macroglobulinemia.
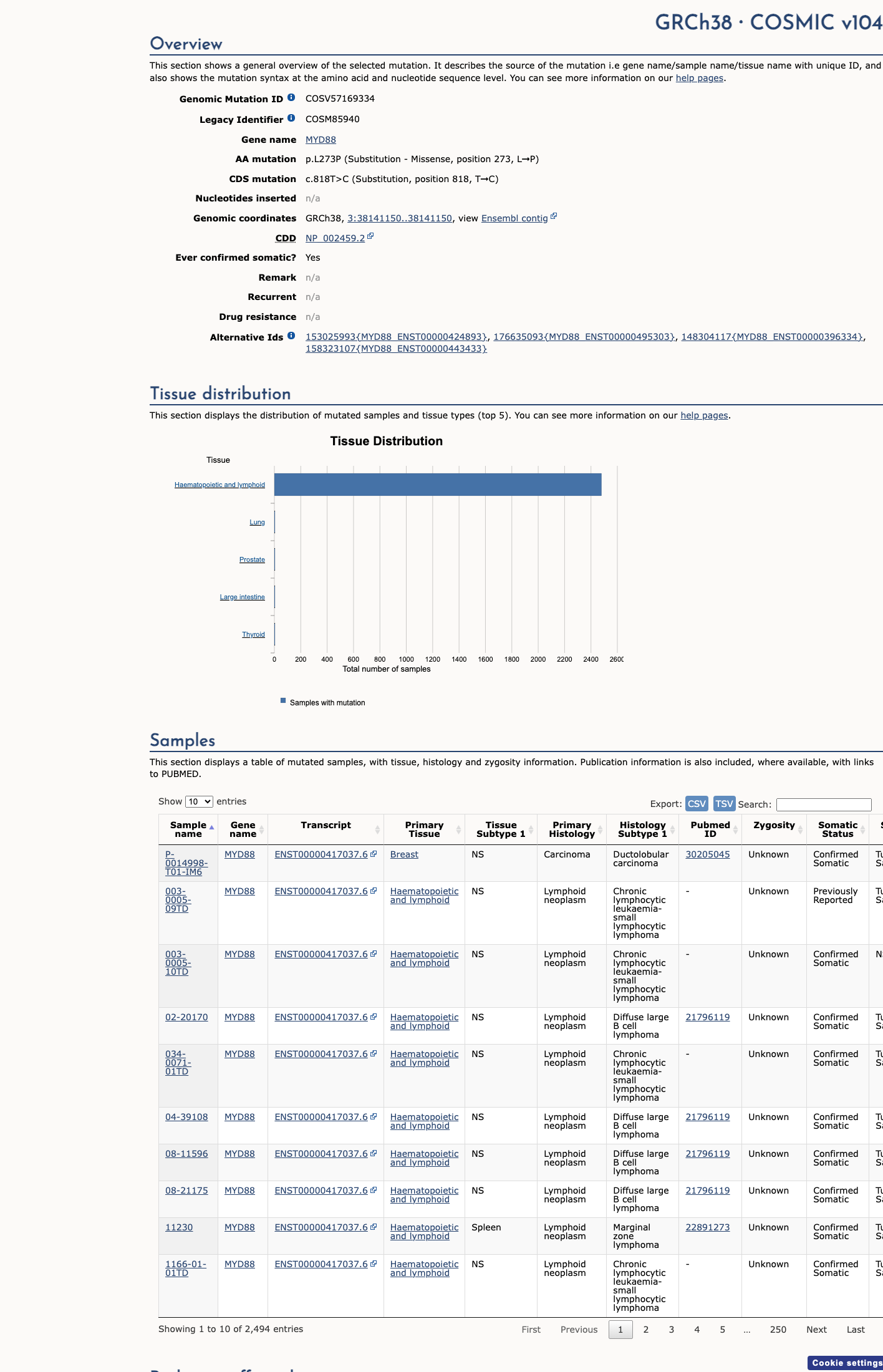
COSMIC
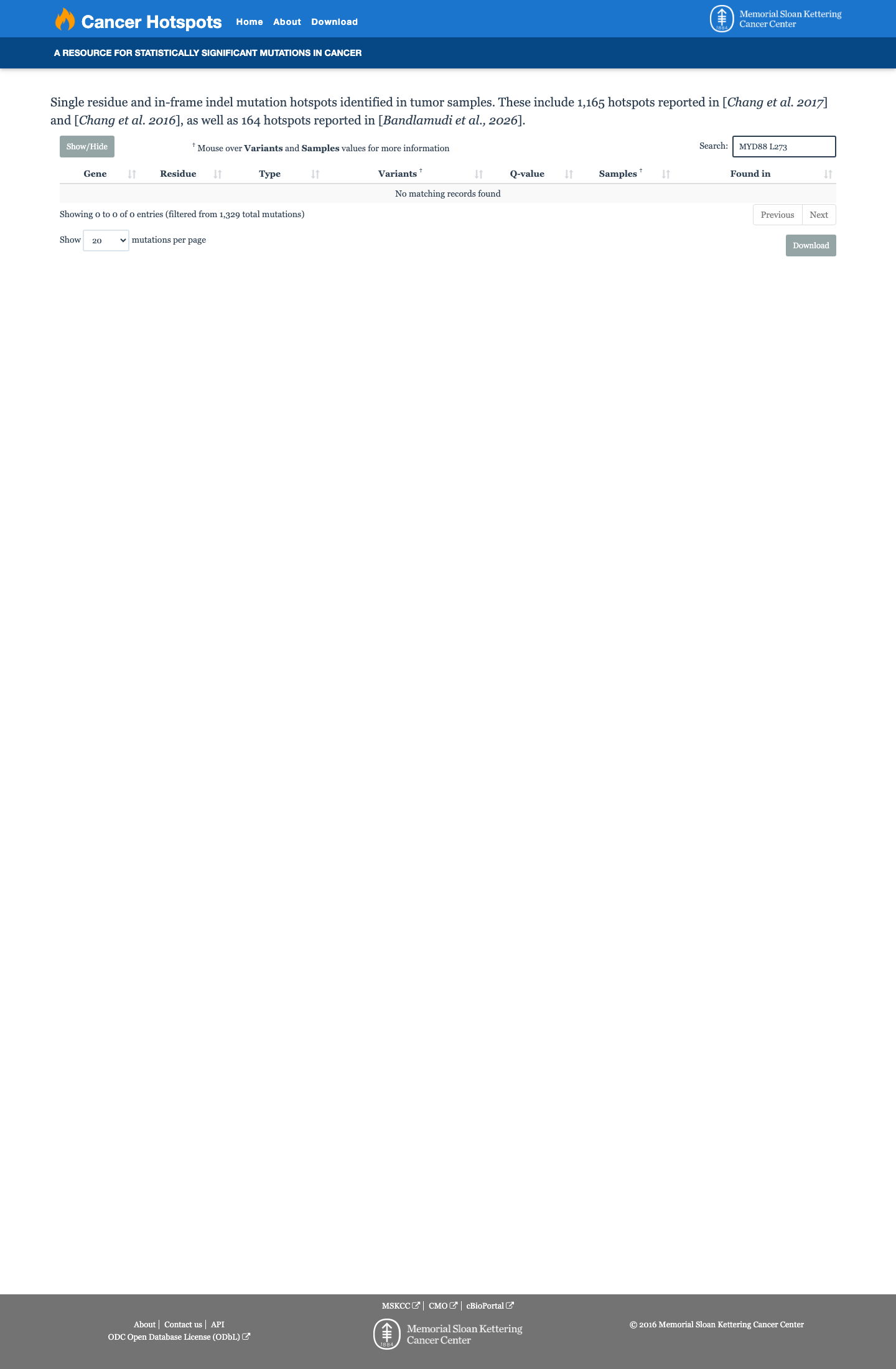
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV57169334, n = 2493 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
5papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 5 further PMIDs triaged but not cited — see Sources & References.
Oncogenically active MYD88 mutations in human lymphoma.
Searched
c.818T>Cp.Leu273ProL265PL273P
Found
MYD88 L265P (Leu273Pro) was identified as a recurrent somatic mutation in activated B-cell-like diffuse large B-cell lymphoma (ABC DLBCL), constitutively activating NF-kappaB and JAK/STAT signaling. Knockdown of MYD88 selectively killed L265P-mutant ABC DLBCL cell lines but not wild-type cells.
Variant
✓ Names this variant
Applied to
→PS3 supports · met
Why
Demonstrated gain-of-function pathogenic mechanism; primary functional evidence for PS3_strong. Also assessed under BS3 where damaging effect contradicts benign interpretation.
MYD88 L265P somatic mutation in Waldenström's macroglobulinemia.
Searched
c.818T>Cp.Leu273ProL265PL273P
Found
MYD88 L265P was detected in over 90% of Waldenstrom macroglobulinemia (WM) cases by whole-genome sequencing of lymphoplasmacytic cells from 30 patients. NF-kappaB activation was confirmed in primary WM cells harboring the mutation.
Variant
✓ Names this variant
Applied to
→PS3 supports · met
→PS4 supports · met
Why
Supported PS3_strong through confirmation of NF-kappaB activation in primary cells, and PS4_strong by establishing high prevalence in affected individuals.
MYD88 L265P somatic mutation in IgM MGUS.
Searched
c.818T>Cp.Leu273ProL265PL273P
Found
MYD88 L265P was identified in IgM-MGUS and shown to localize to the TIR domain BB-loop, a critical functional motif for MYD88 signal transduction and a known mutational hotspot in hematologic malignancies.
Variant
✓ Names this variant
Applied to
→PM1 supports · met
→PS4 supports · met
Why
Supported PS4_strong by documenting L265P in IgM-MGUS precursor state, and PM1_moderate by confirming location in the TIR domain BB-loop mutational hotspot.
Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms.
Searched
c.818T>Cp.Leu273ProL265PL273P
Found
MYD88 L265P was detected in 96% of Waldenstrom macroglobulinemia cases and 54% of IgM monoclonal gammopathy of undetermined significance (IgM-MGUS) cases by allele-specific PCR, establishing L265P as a highly prevalent and early mutational event in WM pathogenesis.
Variant
✓ Names this variant
Applied to
→PS4 supports · met
Why
Strengthened PS4_strong by expanding the known prevalence of L265P in WM and demonstrating its presence in pre-malignant IgM-MGUS.
A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia.
Searched
c.818T>Cp.Leu273ProL265PL273P
Found
MYD88 L265P activated BTK and NF-kappaB signaling in lymphoplasmacytic cells. Pharmacologic inhibition of BTK with ibrutinib selectively reduced survival of L265P-expressing cells, indicating dependence on the MYD88-BTK signaling axis.
Variant
✓ Names this variant
Applied to
→PS3 supports · met
Why
Provided independent functional confirmation of gain-of-function effect and therapeutic vulnerability; referenced in PS3_strong. Also assessed under BS3 where damaging effect contradicts benign interpretation.
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
35101336 ↗
Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC).
CLINVAR
22918138 ↗
Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology.
CLINVAR
34131312 ↗
Chromosomal microarray analysis, including constitutional and neoplastic disease applications, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR
23619274 ↗
American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR