NM_000051.4:c.9139C>T (p.Arg3047Ter) is a nonsense variant in exon 63 of ATM, creating a premature termination codon at the most C-terminal residue considered pathogenic by the HBOP VCEP (p.Arg3047). PVS1 is applied at very strong strength under the ATM PVS1 Decision Tree as a null variant in a gene where loss of function is a known disease mechanism.1 This variant has been reported in ClinVar as Pathogenic by the ClinGen HBOP VCEP expert panel and by 18 clinical laboratories (ClinVar Variation ID: 3029).2 The variant is present in gnomAD v4.1 at an extremely low frequency (AF=1.61×10⁻⁵, 26/1,613,988 alleles; no homozygotes), consistent with a rare disease-causing variant. It is also present in gnomAD v2.1 at similar frequency (AF=1.77×10⁻⁵, 5/282,840 alleles).3 Functional characterization by Guo et al. (2010, PMID 21150274) demonstrates that the R3047X mutant protein is expressed but specifically deficient in oxidation-dependent ATM activation while retaining MRN/DNA-dependent activation. Cells from an A-T patient with this variant showed moderate radiosensitivity, consistent with preserved DNA damage response but lost oxidative stress signaling.4 Chessa et al. (2009, PMID 19691550) identified c.9139C>T in two Italian A-T families from Central Italy, confirming observation of this variant in individuals with classical ataxia-telangiectasia.5 No VCEP-approved functional assay data, case-control studies, or detailed co-segregation data were available from the reviewed sources. Additional supporting evidence may exist in curated databases not accessible in this assessment. The ClinGen expert panel classification of Pathogenic likely incorporates proband data and/or additional supporting criteria not available here.
ATM
Final classification
VUS
ATM c.9139C>T · p.Arg3047Ter
ATM
NM_000051.4:c.9139C>T (p.Arg3047Ter) is a nonsense variant in exon 63 of ATM, creating a premature termination codon at the most C-terminal residue considered pathogenic by the HBOP VCEP (p.Arg3047). PVS1 is applied at very strong strength under the ATM PVS1 Decision Tree as a null variant in a gene where loss of function is a known disease mechanism.
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework was evaluated deterministically with applied criteria: PVS1 very strong, PP5 supporting; no rule matched the adjudicated criteria.
Classification rationale
PVS1PP5
VUS
ATM c.9139C>T
PVS1 + PP5
→
VUS
1
vcep_atm_pvs1_1_5pvs1_gene_context
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 10 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
review
Pathogenic
NM_000051.4:c.9139C>T is a nonsense variant producing p.Arg3047Ter in ATM, a gene where loss of function is a well-established disease mechanism. The VCEP explicitly identifies p.Arg3047 as the most C-terminal residue considered pathogenic (PMIDs: 8755918, 19691550, 18560558, 10980530, 26628246). This null variant truncates the ATM protein within the FATC domain (exon 63, the terminal coding exon). Per the ATM PVS1 Decision Tree, PVS1 is applied at very strong strength.
Nonsense variant (p.Arg3047Ter) in exon 63 of 63 — a null variant in a gene where LOF is a known disease mechanismVCEP PVS1 document confirms p.R3047 is the most C-terminal pathogenic residue in ATMVCEP PVS1 document confirms all ATM exons are considered constitutive without major alternate splice isoforms
Assessed · not applied
Pathogenic
PS3
No variant-specific functional data from VCEP-approved assays (kinase activity assay per Mitui 2009/Barone 2009/Scott 2002, or radiosensitivity assay) are available for c.9139C>T.
PS4
VCEP PS4 requires a case-control study with p≤0.05 AND (OR/HR/RR ≥2 OR lower 95% CI ≥1.5).
PM2
gnomAD v4.1 allele frequency is 1.61×10⁻⁵ (0.00161%, 26/1,613,988 alleles), which exceeds the VCEP PM2_Supporting threshold of ≤0.001% (≤1.0×10⁻⁵).
PP1
PMID 19691550 (Chessa et al.
PP3
PP3 under the ATM VCEP applies to missense variants with REVEL >0.7333 or to splicing variants with SpliceAI ≥0.2.
Benign
BA1
VCEP BA1 threshold is grpmax Filtering AF >0.5% in gnomAD v4.
BS1
VCEP BS1 threshold is grpmax Filtering AF >0.05% in gnomAD v4.
BS3
No variant-specific benign functional data from VCEP-approved assays are available.
BP2
BP2 requires proband-level data (confirmed in trans with a pathogenic variant in unaffected individuals ≥18 years with no evidence of A-T) per the ATM PM3/BP2 table.
BP4
BP4 under the ATM VCEP applies to missense variants with REVEL ≤0.249 or splicing variants with SpliceAI ≤0.1.
N/A · 16
PS1 · PS2 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP4 · BS2 · BS4 · BP1 · BP3 · BP5 · BP6 · BP7
Research & evidence
Population frequency

gnomAD v4.1
gnomAD v2.1
v4.1
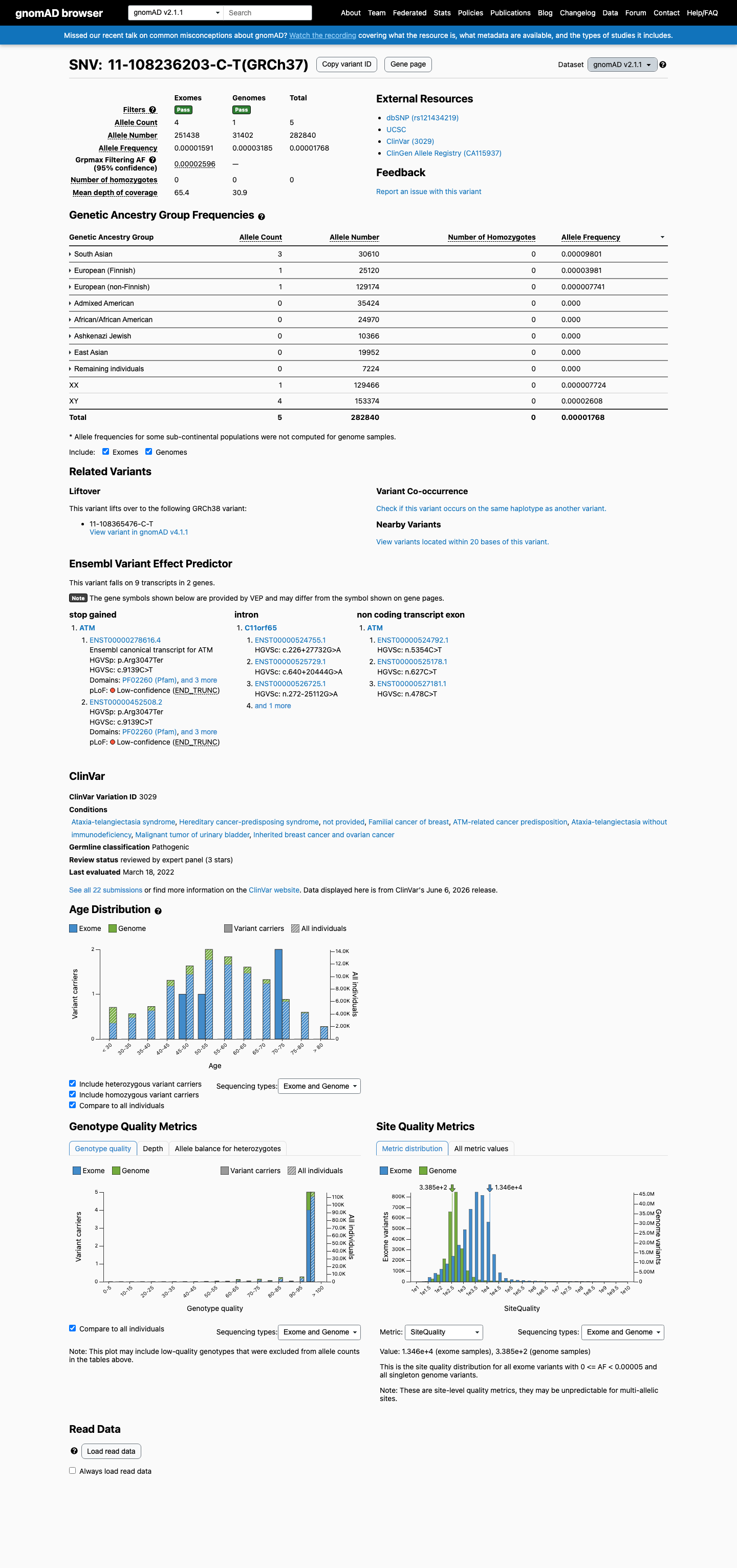
This variant is present in gnomAD v4.1 (AF= 1.61092e-05; MAF= 0.00161%, 26/1613988 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 8.78368e-05; MAF= 0.00878%, 8/91078 alleles, homozygotes = 0); grpmax FAF= 4.368e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 1.76778e-05; MAF= 0.00177%, 5/282840 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 9.80072e-05; MAF= 0.00980%, 3/30610 alleles, homozygotes = 0); grpmax FAF= 2.596e-05.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0016%
· 26 / 1,613,988
0 hom · FAF 0.0044%
0 hom · FAF 0.0044%
South Asian 8 / 91,078 |
0.0088% |
European (Finnish) 2 / 64,002 |
0.0031% |
African/African American 1 / 74,912 |
0.0013% |
European (non-Finnish) 15 / 1,180,030 |
0.0013% |
+ 6 not observed (Remaining individuals, Admixed American, Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0018%
· 5 / 282,840
0 hom · FAF 0.0026%
0 hom · FAF 0.0026%
South Asian 3 / 30,610 |
0.0098% |
European (Finnish) 1 / 25,120 |
0.004% |
European (non-Finnish) 1 / 129,174 |
0.00077% |
+ 5 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
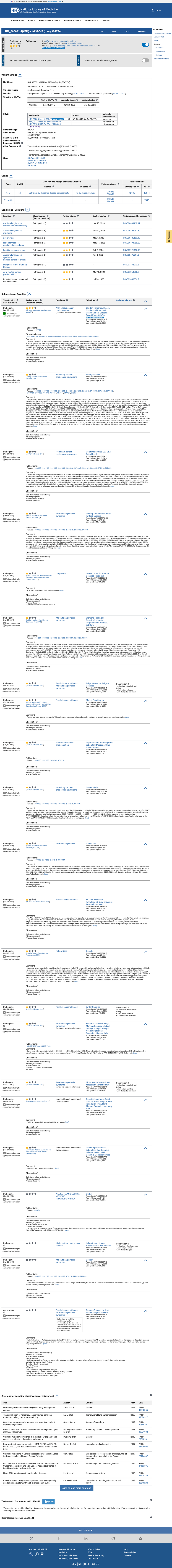
This variant has been reported in ClinVar as Pathogenic (18 clinical laboratories) and as Pathogenic by ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Variant Curation Expert Panel, ClinGen (expert panel). (ClinVarID = 3029)
In silico
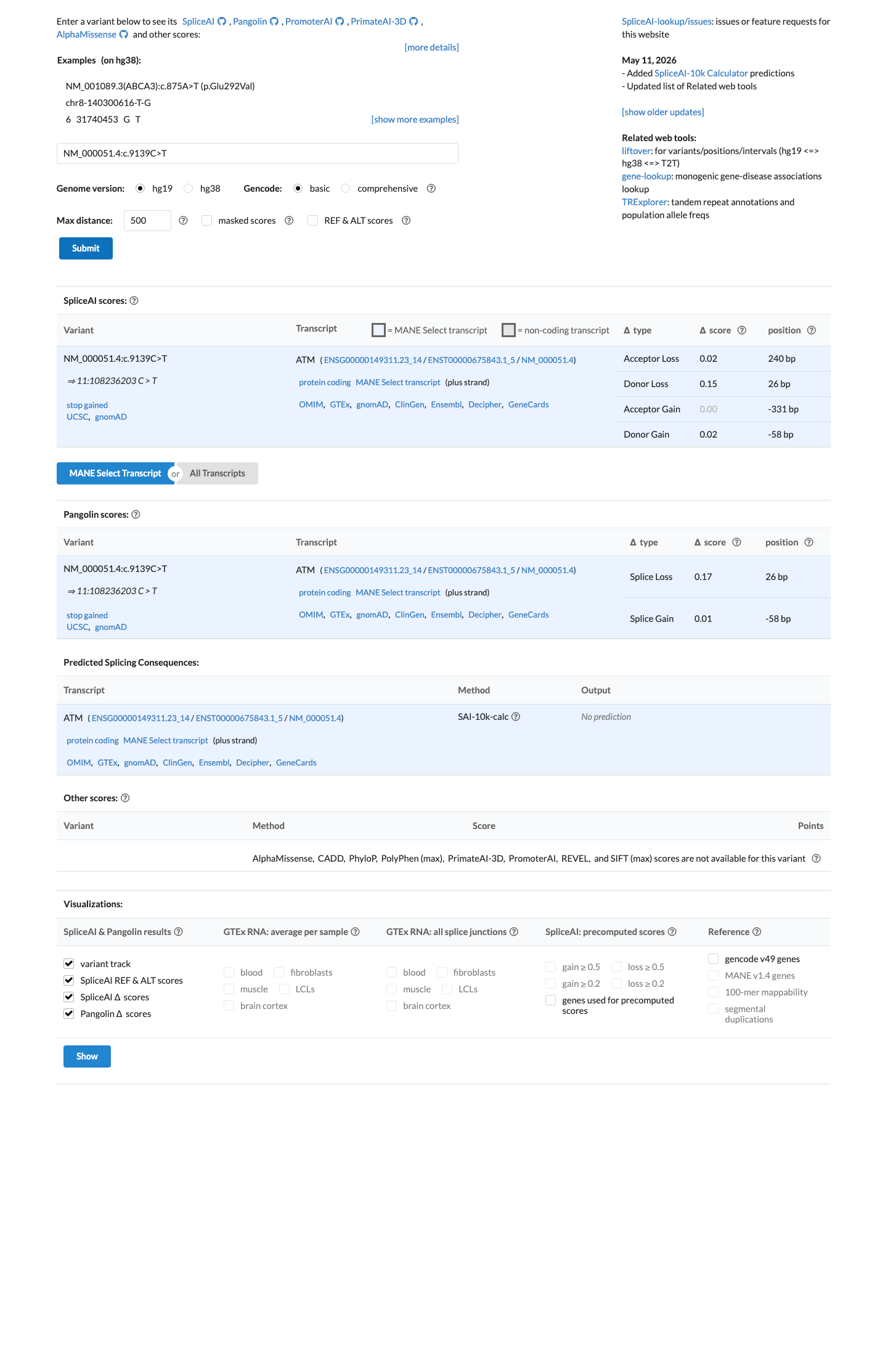
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.15). BayesDel score = 0.61701.
Functional
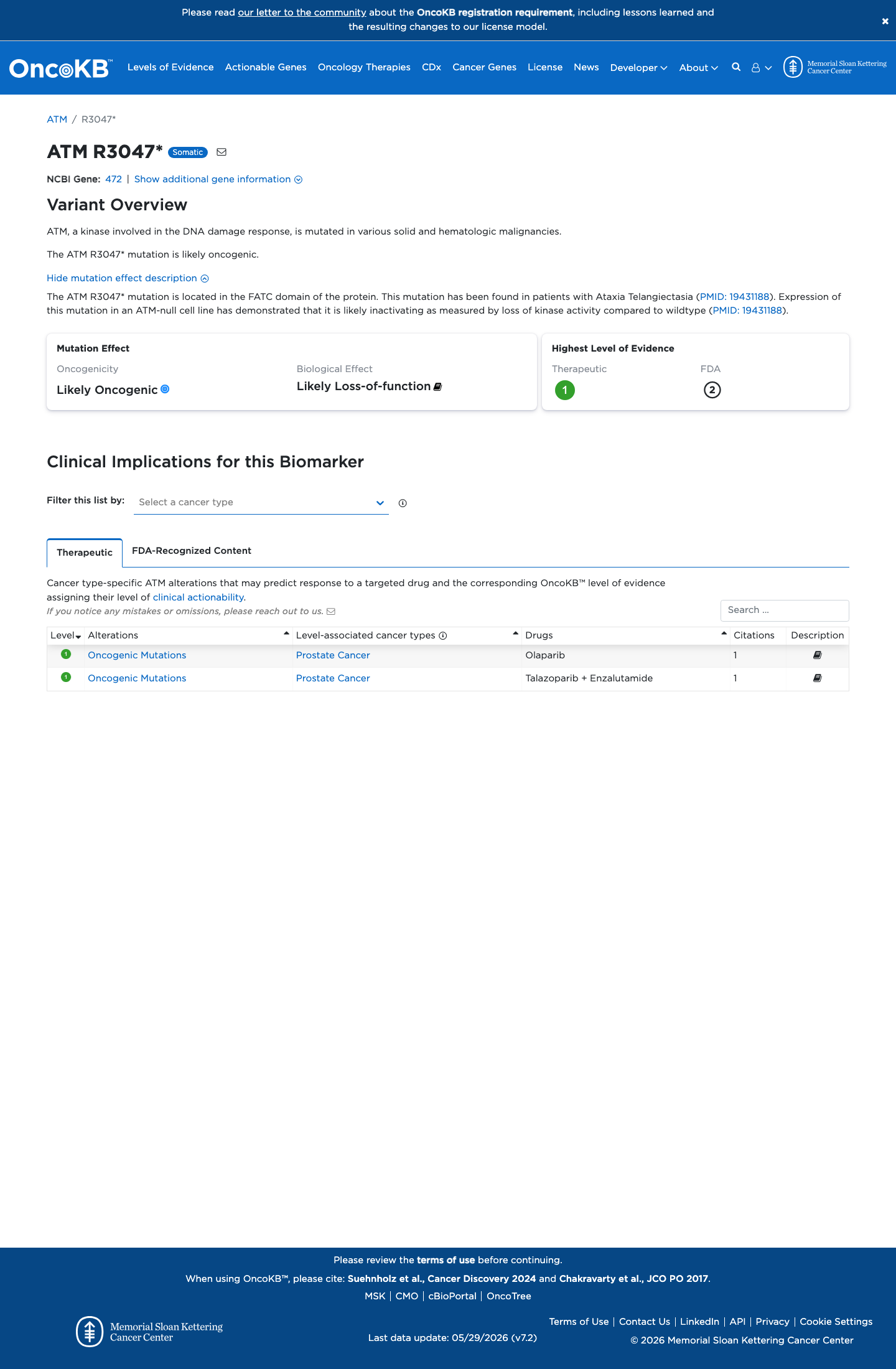
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53724126, n = 18 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 5 further PMIDs triaged but not cited — see Sources & References.
Founder effects for ATM gene mutations in Italian Ataxia Telangiectasia families.
Searched
c.9139C>Tp.Arg3047TerR3047*9139C>T
Found
Two Italian A-T families from Central Italy carried the c.9139C>T mutation. Haplotype analysis was performed across 48 A-T families with 16 recurrent mutations, including c.9139C>T. The variant was identified as one of several recurrent ATM mutations with founder effects in the Italian population.
Variant
✓ Names this variant — characterised directly
Applied to
→PVS1 supports · met
Why
Confirms variant observation in A-T patients. No detailed co-segregation data (number of affected relatives, genotypes) was available to support PP1. Referenced in PVS1 assessment as corroborating the pathogenic status of p.R3047.
Two families carrying the c.9139C>T mutation and two with c.4396C>T all originated from Central Italy.
Location Results section, line 235-236 · Context Mutation screening (DHPLC, PTT, SSCP, HA, REF) of 104 Italian A-T patients from 91 families; haplotype analysis with 5 microsatellite markers. · full text
ATM activation in the presence of oxidative stress.
Searched
c.9139C>Tp.Arg3047TerR3047*R3047X3047
Found
The R3047X mutant ATM protein (lacking the C-terminal 10 amino acids) can be fully activated by MRN and DNA but cannot be activated by oxidation in vitro. Cells from an A-T patient expressing R3047X were specifically deficient in oxidative activation of ATM but showed normal ATM activation in response to DNA damage, with only moderate radiosensitivity. The variant is described as a causative mutation in several A-T patients, sometimes classified as A-T variants due to milder phenotype.
Variant
✓ Names this variant — characterised directly
Applied to
→PVS1 supports · met
Why
Variant-specific functional data confirms R3047X protein is expressed but has selective loss of oxidation-dependent ATM activation. The assay is not among the VCEP-approved functional assays (kinase activity per Mitui 2009/Barone 2009/Scott 2002), so it does not independently meet PS3. Referenced in PVS1 assessment to support functional significance of the C-terminal truncation despite potential NMD escape.
a mutant lacking the last ten amino acids of the ATM C-terminus results in a mutant protein (R3047X, Fig. 1A) that can be fully activated by MRN and DNA but cannot be activated by oxidation in vitro.
Location Results section; Figure 1A · Context In vitro kinase assays with purified ATM wild-type and mutant proteins; phosphorylation of GST-p53 substrate assessed by western blotting. Patient-derived cells used for cellular validation. · full text
Sources & reference links
9Sources
Triaged references · 5 PMIDs not cited in assessment
19431188 ↗
Modeling ATM mutant proteins from missense changes confirms retained kinase activity.
ONCOKB
10980530 ↗
Characterization of ATM mutations in 41 Nordic families with ataxia telangiectasia.
CLINVAR
18560558 ↗
Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia.
CLINVAR
22649200 ↗
Classical ataxia telangiectasia patients have a congenitally aged immune system with high expression of CD95.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR