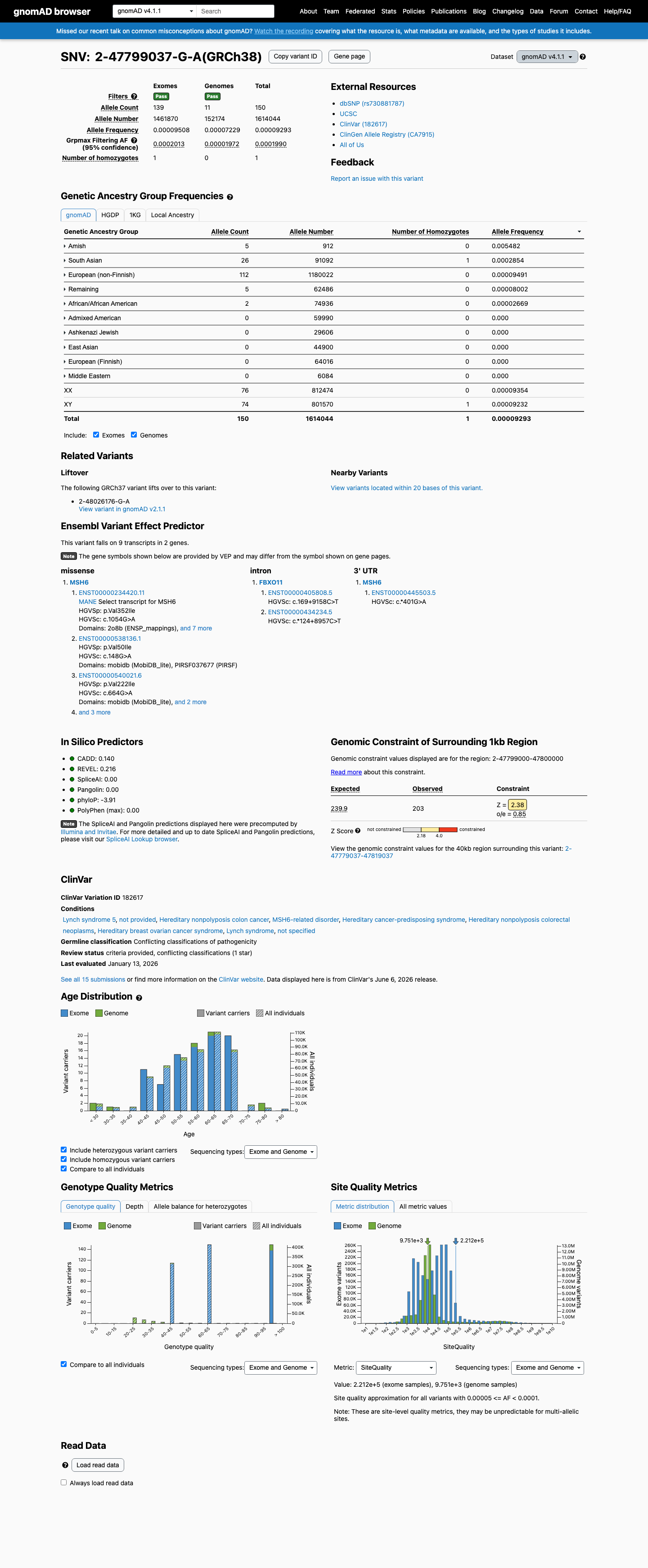
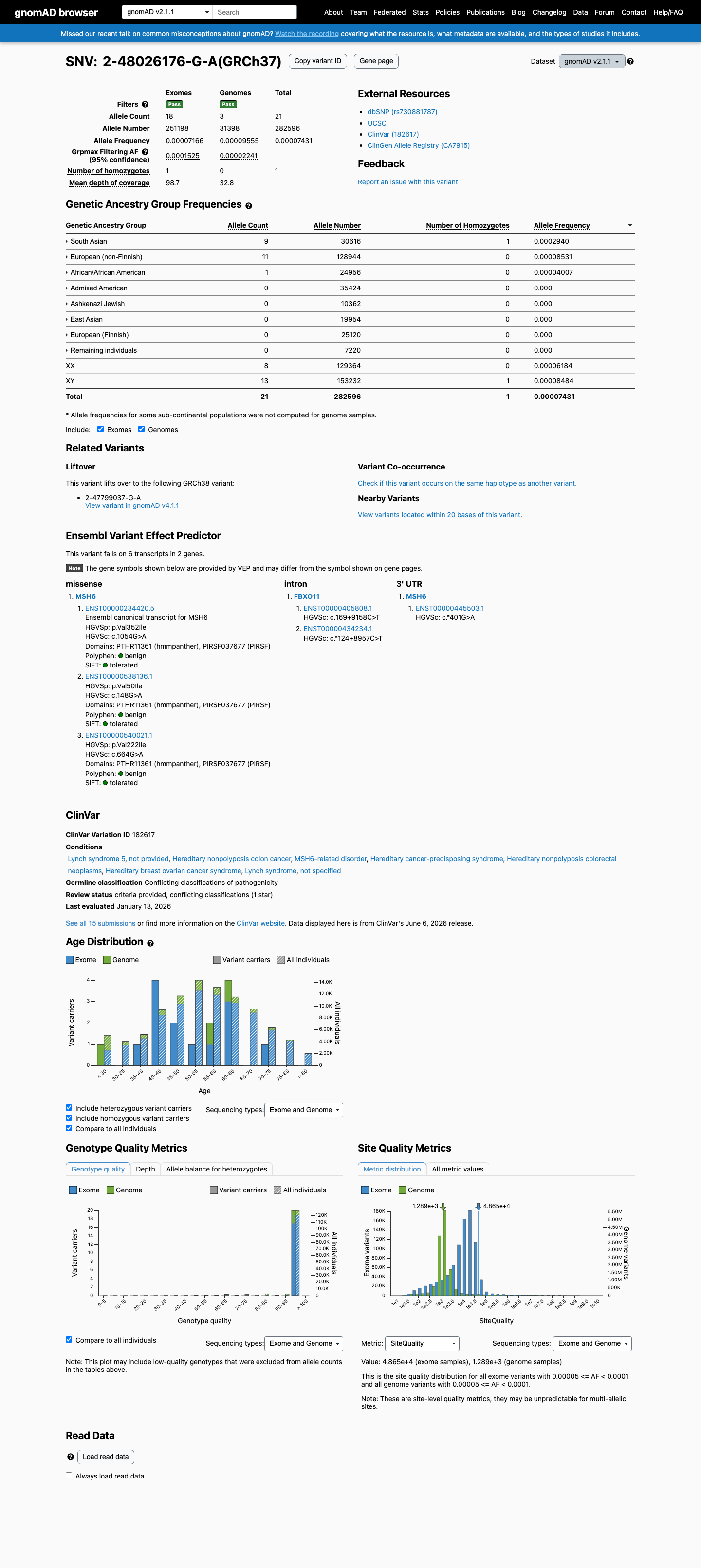
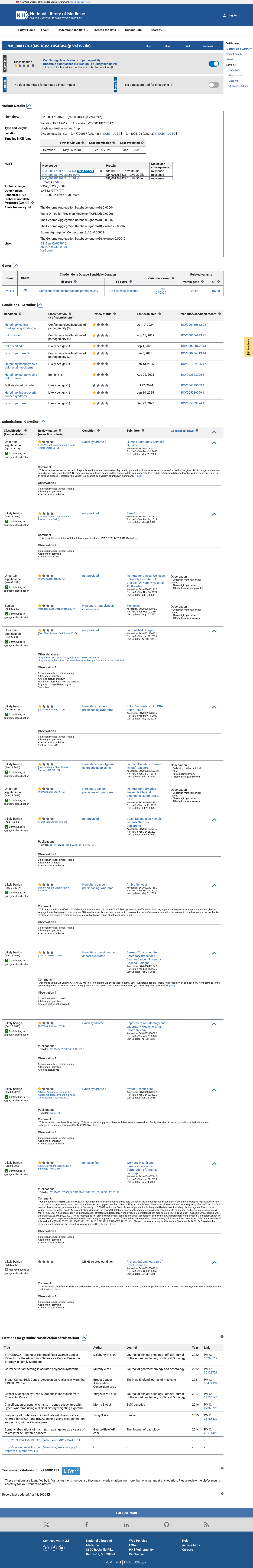
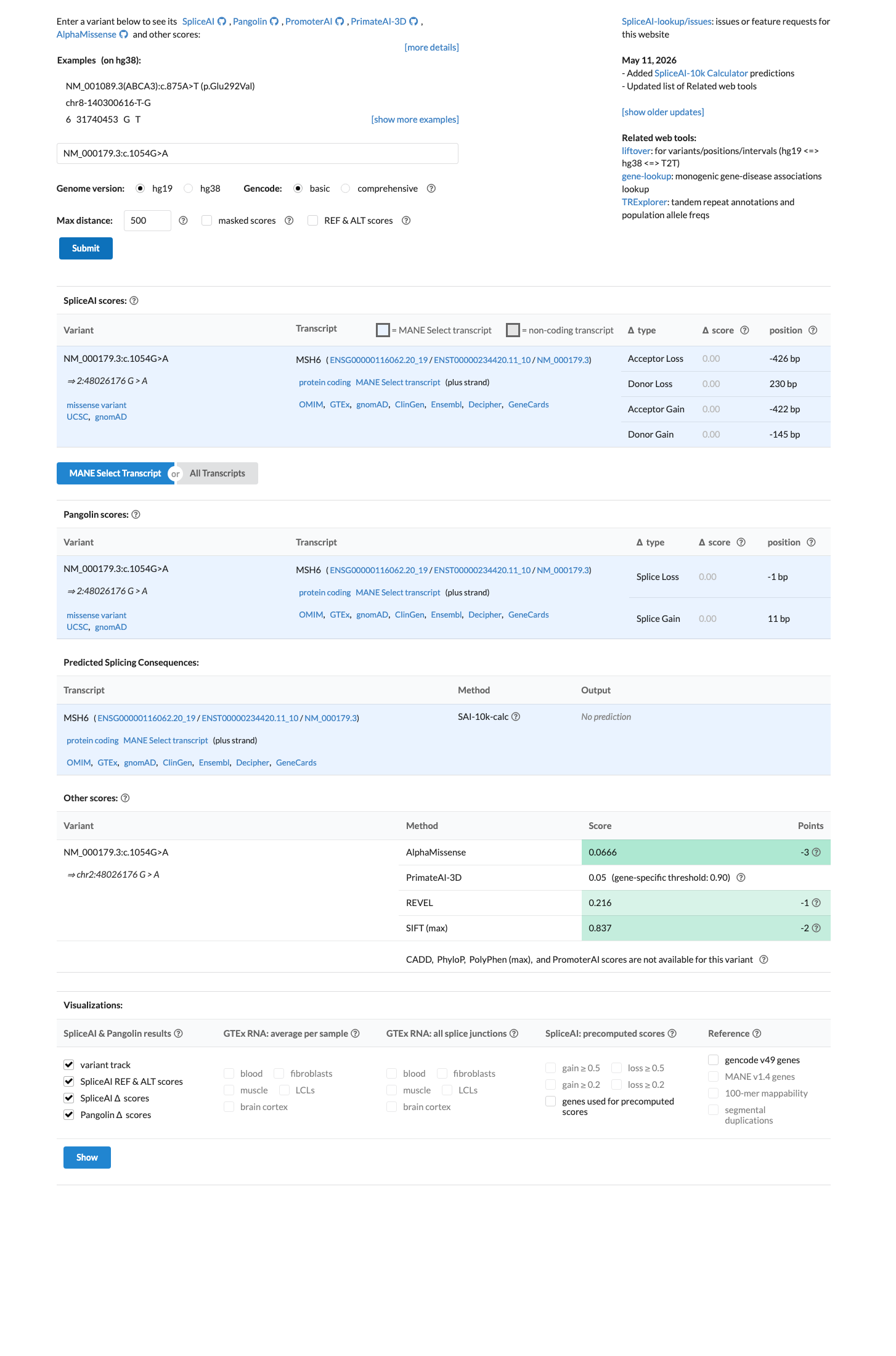
NM_000179.3(MSH6):c.1054G>A (p.Val352Ile) is a missense substitution in exon 4 of MSH6. This variant has been observed in gnomAD v4.1 at an overall allele frequency of 0.000093 (150/1,614,044 alleles, 1 homozygote), with a grpmax filtering allele frequency of 0.000199 (0.0199%). In gnomAD v2.1 the variant is present at an allele frequency of 0.000074 (21/282,596 alleles, 1 homozygote). It is absent from gnomAD-Canada v1.0.1 The gnomAD v4.1 grpmax FAF (0.000199) is below the InSiGHT MSH6 VCEP BS1 threshold of ≥0.00022 and far below the BA1 threshold of ≥0.0022, so neither allele-frequency-based benign criterion is met. The overall allele frequency also exceeds the PM2 supporting threshold of <0.00002, so PM2 is not met.2 Computational predictors strongly favor a benign effect: the HCI prior probability for pathogenicity is 0.0007 (far below the BP4_Supporting threshold of <0.11), SpliceAI predicts no splicing impact (max delta 0.00), REVEL score is 0.216, and BayesDel score is -0.443.3 The variant was detected as a somatic finding in a tumor from a suspected Lynch syndrome patient (PMID:25111426), where it was classified as class 2 (likely not pathogenic) with 34% variant allele frequency, retention of heterozygosity, and normal MMR protein expression by IHC. This somatic observation is consistent with a benign or low-impact variant but does not independently meet any VCEP criterion in a germline context.4 In ClinVar, this variant is reported as Likely benign by 9 clinical laboratories, Uncertain significance by 4, and Benign by 1 (ClinVar Variation ID: 182617). The InSiGHT expert panel has not issued a classification for this variant.5 No pathogenic or likely pathogenic missense variants at the same amino acid residue (Val352) were identified in InSiGHT VCEP records, so PM5 cannot be assessed.6 Applying the InSiGHT MSH6 VCEP version 2.0.0 combining rules: BP4_Supporting is the only criterion met. With a single benign supporting criterion, neither the Likely Benign threshold (≥2 benign supporting per Rule 19, or 1 benign strong + 1 benign supporting per Rule 18) nor any pathogenic threshold is reached. The variant is classified as Uncertain Significance.
MSH6
Final classification
VUS
MSH6 c.1054G>A · p.Val352Ile
MSH6
NM_000179.3(MSH6):c.1054G>A (p.Val352Ile) is a missense substitution in exon 4 of MSH6.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MSH6 Version 2.0.0 v2.0.0 criteria-combination framework was evaluated deterministically with applied criteria: BP4 supporting benign; no rule matched the adjudicated criteria.
Classification rationale
BP4
VUS
MSH6 c.1054G>A
BP4
→
VUS
3
hci_priorspliceai ↗revelbayesdel
6
pm5_candidates
Gene diagram
· NM_000179.3 · variants mapped to exon structure
MSH6
NM_000179.3
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 14 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
BP4
supporting
Benign
HCI prior probability for pathogenicity 0.0007 falls below the InSiGHT MSH6 VCEP BP4_Supporting threshold of <0.11. SpliceAI max delta score 0.00 confirms no predicted splicing impact. REVEL score 0.216 and BayesDel score -0.443 are also consistent with a benign computational profile.
HCI prior = 0.0007 (<0.11 threshold for BP4_Supporting). SpliceAI max delta = 0.00. REVEL = 0.216. BayesDel = -0.443.
Assessed · not applied
Pathogenic
PS1
No previously classified pathogenic variant encoding the same amino acid change (Val352Ile) via a different nucleotide change was identified in InSiGHT MSH6 VCEP records.
PS2
No de novo observation data available for this variant.
PS3
No calibrated functional assay data available in the VCEP MMR functional assay documentation.
PM2
gnomAD v4.1 allele frequency 0.000093 (150/1,614,044 alleles) exceeds the InSiGHT MSH6 VCEP PM2 threshold of <0.00002 (<1 in 50,000 alleles).
PM5
No pathogenic or likely pathogenic missense variant at the same amino acid residue (Val352) identified in InSiGHT MSH6 VCEP records.
PP1
No co-segregation data available for this variant.
PP3
HCI prior probability for pathogenicity 0.0007 is far below the InSiGHT MSH6 VCEP PP3_Moderate threshold (>0.81) and PP3_Supporting threshold (>0.68).
PP4
No tumor phenotype data (MSI status, MMR IHC) specific to this germline variant available in evidence sources.
Benign
BA1
gnomAD v4.1 grpmax filtering allele frequency 0.000199 (0.0199%) is below the InSiGHT MSH6 VCEP BA1 threshold of ≥0.0022 (0.22%).
BS1
gnomAD v4.1 grpmax filtering allele frequency 0.000199 (0.0199%) is slightly below the InSiGHT MSH6 VCEP BS1 threshold of ≥0.00022 (0.022%).
BS2
No trans co-occurrence data with a known pathogenic MSH6 variant available.
BS3
No calibrated functional assay data available for this variant in the VCEP MMR functional assay documentation.
BS4
No co-segregation data available for this variant.
BP5
No tumor phenotype data (MSS/MSI status, MMR IHC) from germline carriers of this variant available.
N/A · 13
PVS1 · PS4 · PM1 · PM3 · PM4 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 9.29343e-05; MAF= 0.00929%, 150/1614044 alleles, homozygotes = 1) and has highest observed frequency in the Amish population (AF= 0.00548246; MAF= 0.54825%, 5/912 alleles, homozygotes = 0); grpmax FAF= 0.00019901.
v2.1
This variant is present in gnomAD v2.1 (AF= 7.4311e-05; MAF= 0.00743%, 21/282596 alleles, homozygotes = 1) and has highest observed frequency in the South Asian population (AF= 0.000293964; MAF= 0.02940%, 9/30616 alleles, homozygotes = 1); grpmax FAF= 0.00015255.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00021715526601520088, 4/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0093%
· 150 / 1,614,044
1 hom · FAF 0.02%
1 hom · FAF 0.02%
Amish 5 / 912 |
0.55% |
South Asian 26 / 91,092 |
0.029% 1 hom |
European (non-Finnish) 112 / 1,180,022 |
0.0095% |
Remaining individuals 5 / 62,486 |
0.008% |
African/African American 2 / 74,936 |
0.0027% |
+ 5 not observed (Admixed American, European (Finnish), East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0074%
· 21 / 282,596
1 hom · FAF 0.015%
1 hom · FAF 0.015%
South Asian 9 / 30,616 |
0.029% 1 hom |
European (non-Finnish) 11 / 128,944 |
0.0085% |
African/African American 1 / 24,956 |
0.004% |
+ 5 not observed (Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
0.022%
· 4 / 18,420
0 hom
0 hom
South Asian 1 / 1,362 |
0.073% |
European (non-Finnish) 3 / 11,742 |
0.026% |
+ 7 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals)
ClinVar
This variant has been reported in ClinVar as Likely benign (9 clinical laboratories) and as Uncertain significance (4 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 182617)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.216. BayesDel score = -0.442852. HCI prior probability for pathogenicity = 0.0007. MAPP score = 2.28. Custom PP2 score = 0.001.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MSH6, a DNA mismatch repair protein, is frequently mutated in colorectal, small bowel, and endometrial cancers.
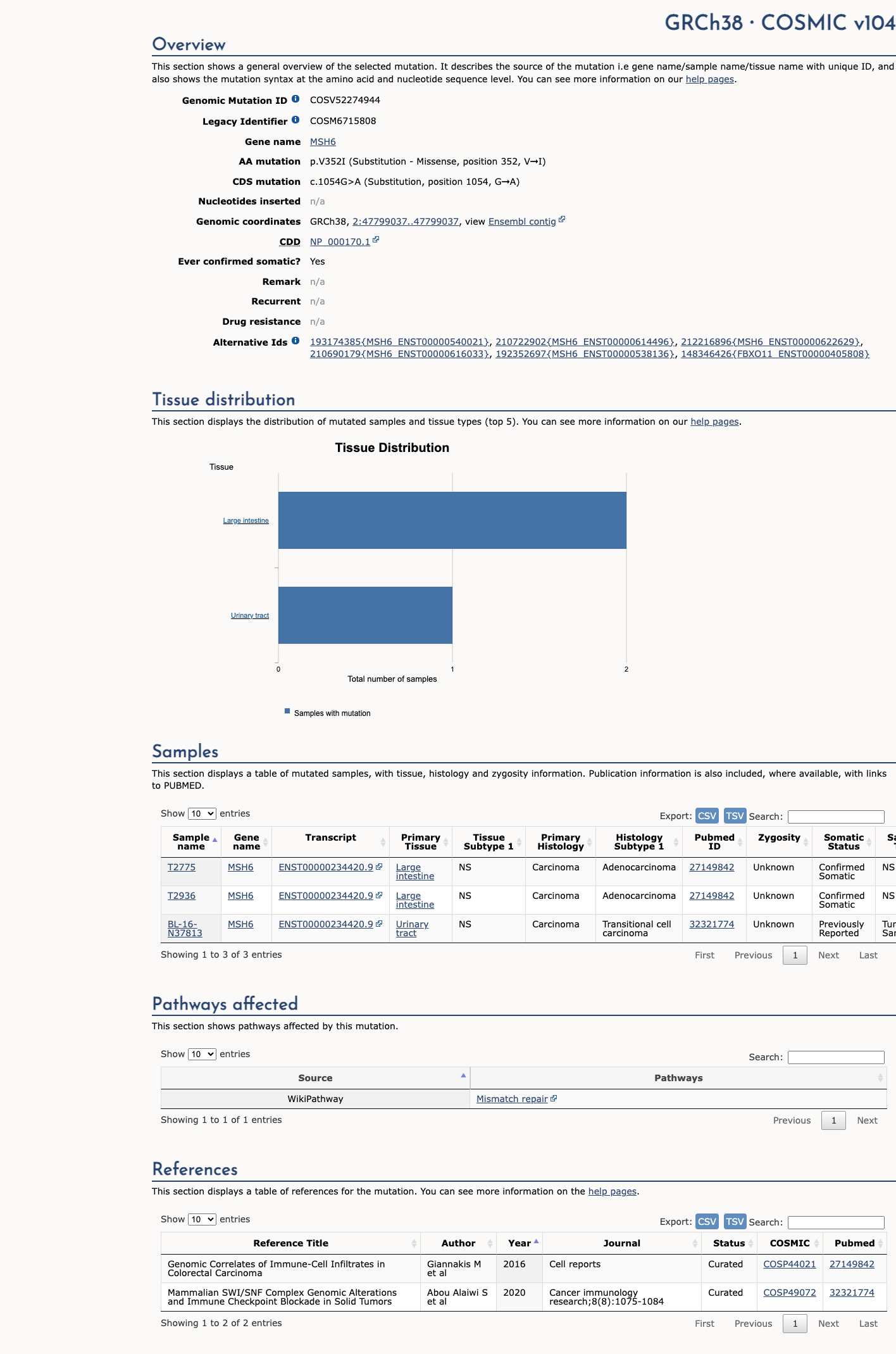
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV52274944, n = 3 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
23535968 ↗
Informing family members of individuals with Lynch syndrome: a guideline for clinical geneticists.
CLINVAR
25111426 ↗
Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers.
CLINVAR
25186627 ↗
Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR