NM_006231.4:c.6748-18G>A is an intronic variant in POLE located 18 bases upstream of exon 48. This variant has been observed at very low frequency in population databases (gnomAD v2.1: 8/249,726 alleles, AF=0.0032%; gnomAD v4.1: 13/1,594,924 alleles, AF=0.00082%) with no homozygotes.1 The variant meets PM2 (Supporting) due to its very low population frequency, below the 0.1% threshold.2 SpliceAI predicts no impact on splicing (max delta score = 0.00), and no other computational tools predict a deleterious effect. PP3 is not met and BP4 does not reach the 'multiple lines' threshold.3 This variant has been reported in ClinVar as Likely benign by Labcorp Genetics (formerly Invitae), meeting BP6 (Supporting).4 The variant is not located in a known POLE mutational hotspot or functional domain. The custom POLE framework (León-Castillo et al. 2020) applies exclusively to missense variants and does not provide applicable PM1, PS4, PP3, or BP4 rules for this intronic variant.5 No functional studies, segregation data, de novo observations, or case-control data are available. PVS1 is not applicable as the variant does not meet null-variant criteria. PS1, PM5, PP2, BP1, and BP7 are not applicable due to the intronic nature of the variant.6 The evidence profile consists of one pathogenic supporting criterion (PM2) and one benign supporting criterion (BP6), resulting in a variant of uncertain significance (VUS) under the custom POLE framework using generic ACMG/AMP 2015 combination rules.7
POLE
Final classification
VUS
POLE c.6748-18G>A · p.?
POLE
NM_006231.4:c.6748-18G>A is an intronic variant in POLE located 18 bases upstream of exon 48. This variant has been observed at very low frequency in population databases (gnomAD v2.1: 8/249,726 alleles, AF=0.0032%; gnomAD v4.1: 13/1,594,924 alleles, AF=0.00082%) with no homozygotes.
The adjudicated evidence profile contains one pathogenic supporting criterion (PM2) and one benign supporting criterion (BP6). The León-Castillo et al. 2020 custom POLE gene framework provides explicit ACMG/AMP 2015 combination rules for pathogenic, likely pathogenic, benign, and likely benign. None of the combination paths are satisfied by this evidence profile: PM2_Supporting alone does not reach any pathogenic or likely pathogenic threshold (which require at minimum PVS1+Moderate, Strong+Moderate, or 3 Moderate), and BP6_Supporting alone does not reach the likely benign threshold (which requires either 1 Strong benign + 1 Supporting benign or ≥2 Supporting benign). Conflicting pathogenic and benign evidence with no applicable combination rule defaults to VUS.
Classification rationale
PM2
BP6
VUS
POLE c.6748-18G>A
PM2 + BP6
→
VUS
5
vcep_path_250_323
6
pvs1_variant_assessmentpm5_candidates
7
final_classification_framework
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
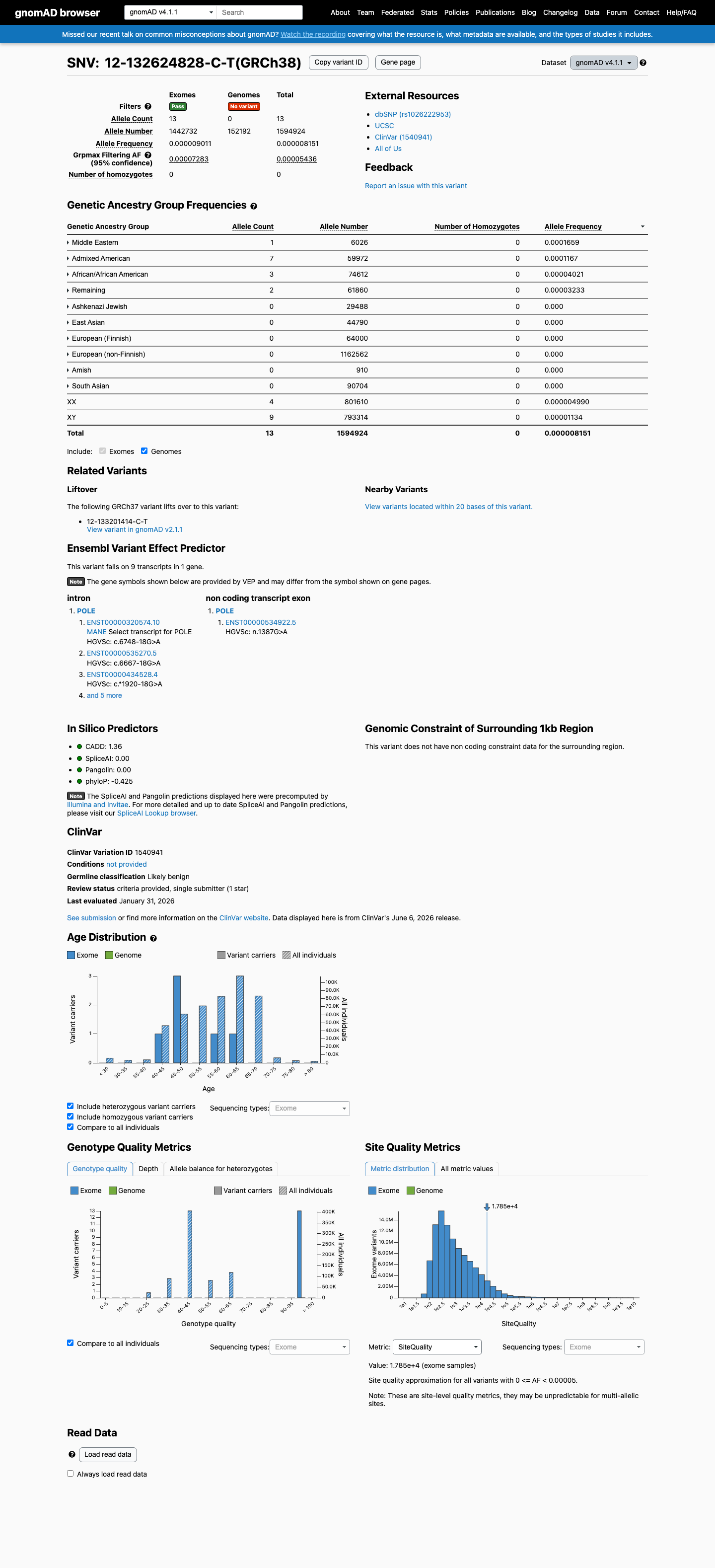
gnomAD v4.1
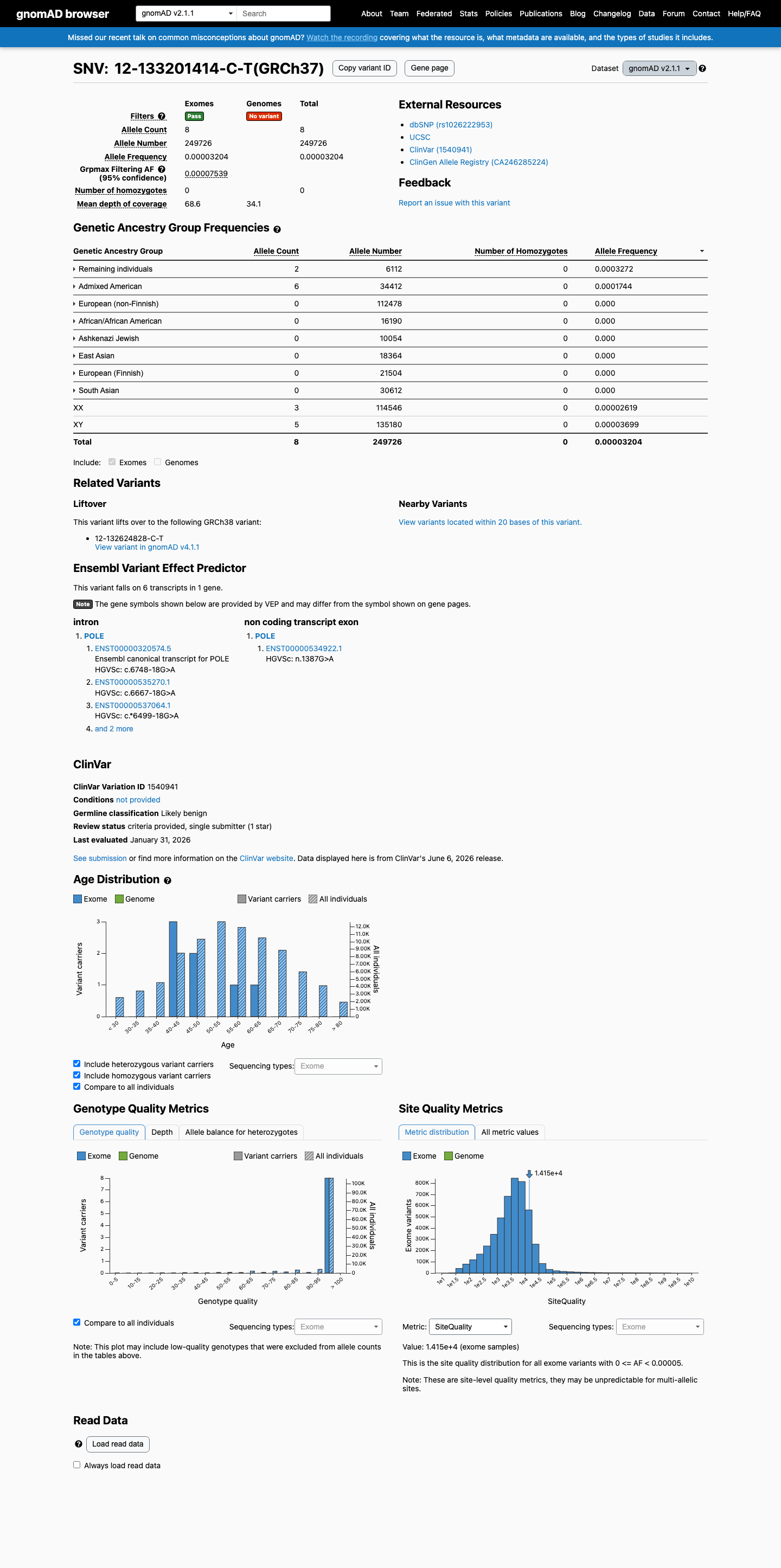
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 8.15086e-06; MAF= 0.00082%, 13/1594924 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000165948; MAF= 0.01659%, 1/6026 alleles, homozygotes = 0); grpmax FAF= 5.436e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.20351e-05; MAF= 0.00320%, 8/249726 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.000327225; MAF= 0.03272%, 2/6112 alleles, homozygotes = 0); grpmax FAF= 7.539e-05.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00082%
· 13 / 1,594,924
0 hom · FAF 0.0054%
0 hom · FAF 0.0054%
Middle Eastern 1 / 6,026 |
0.017% |
Admixed American 7 / 59,972 |
0.012% |
African/African American 3 / 74,612 |
0.004% |
Remaining individuals 2 / 61,860 |
0.0032% |
+ 6 not observed (European (Finnish), Amish, East Asian, South Asian, Ashkenazi Jewish, European (non-Finnish))
gnomAD v2.1
0.0032%
· 8 / 249,726
0 hom · FAF 0.0075%
0 hom · FAF 0.0075%
Remaining individuals 2 / 6,112 |
0.033% |
Admixed American 6 / 34,412 |
0.017% |
+ 6 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
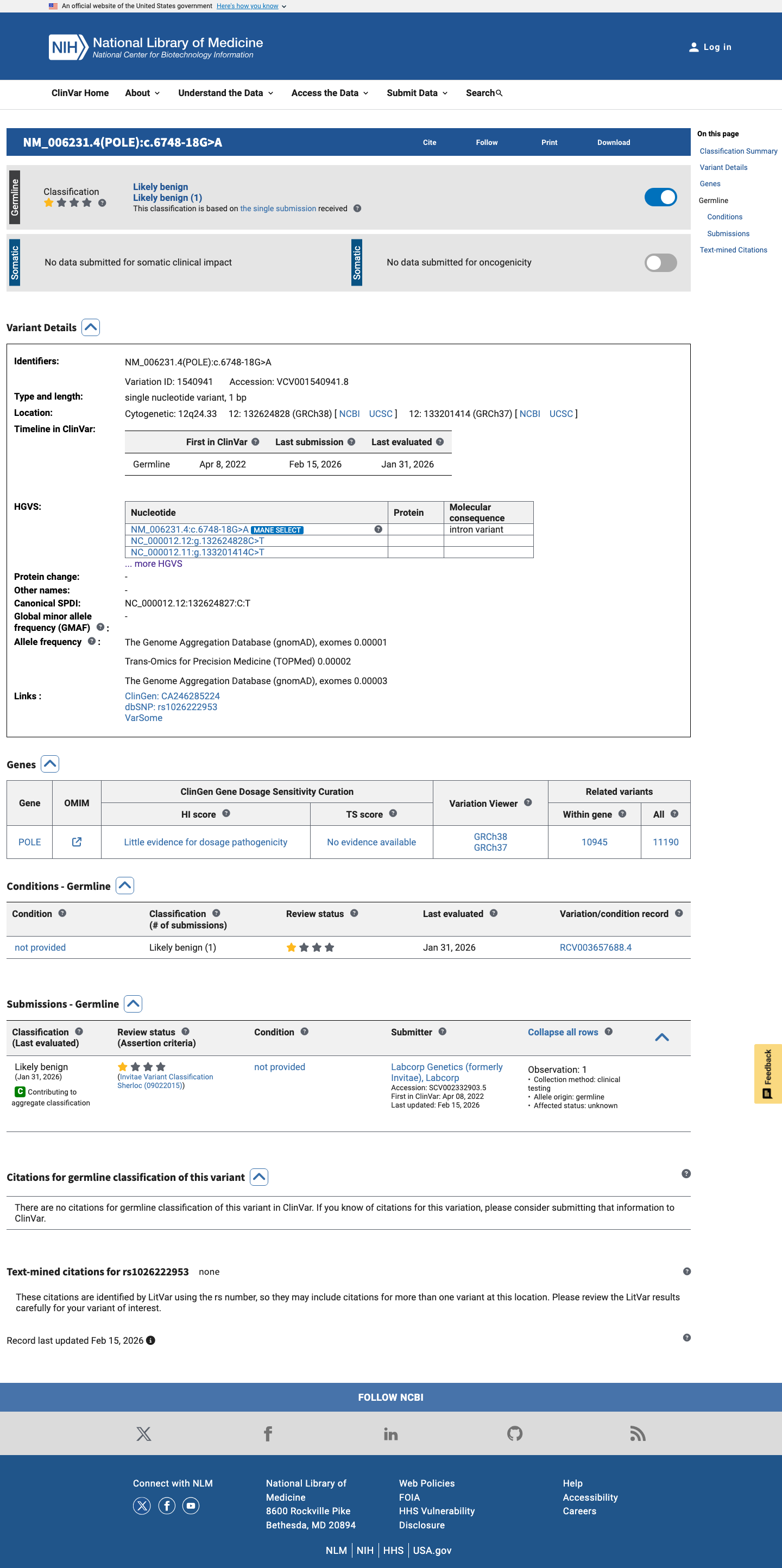
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory). (ClinVarID = 1540941)
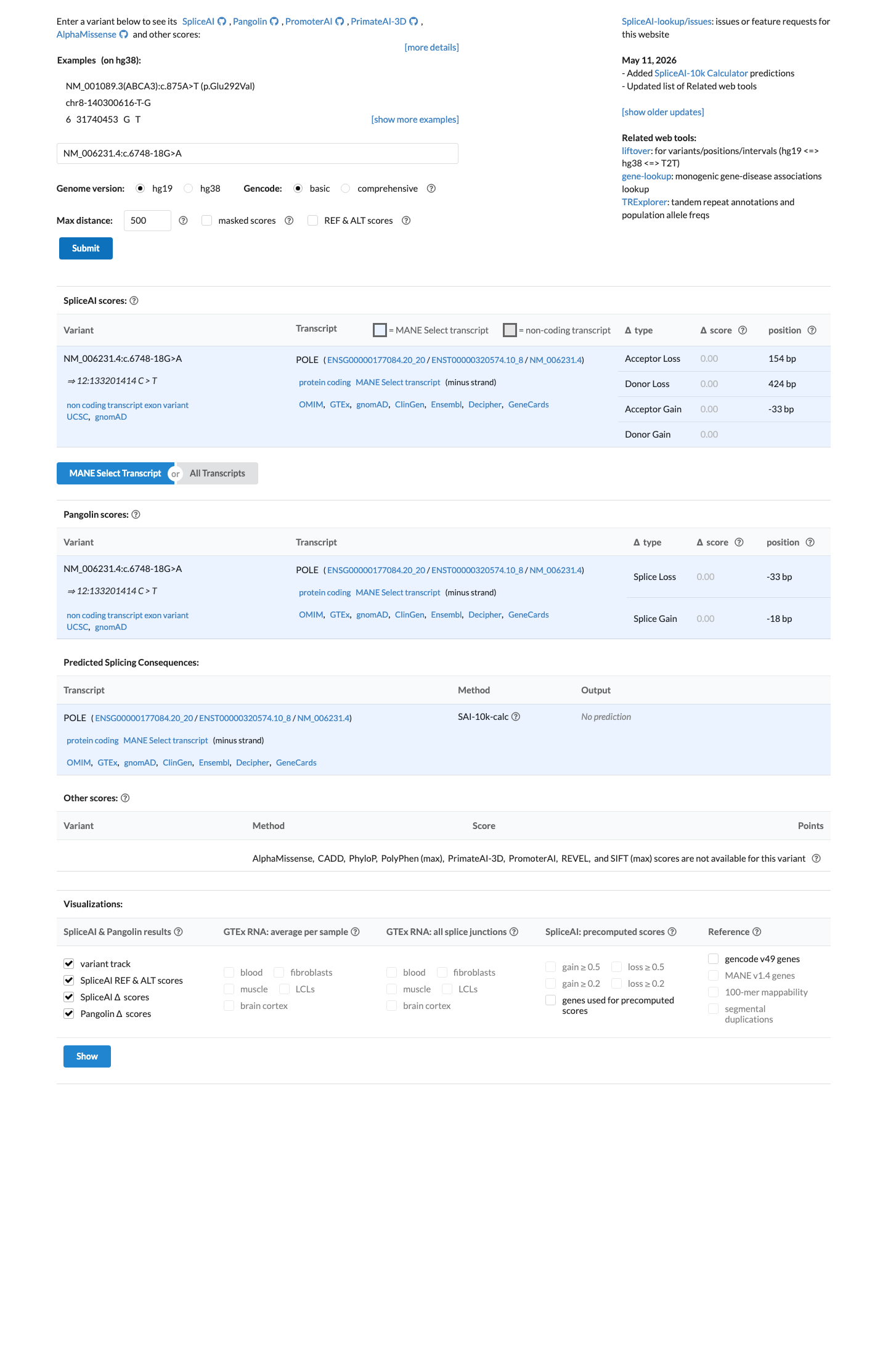
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR