NM_005188.3:c.1096-1G>T is a canonical splice acceptor variant in CBL, disrupting the AG dinucleotide at the intron 7 / exon 8 boundary.1 CBL loss-of-function is an established disease mechanism for a RASopathy phenotype with predisposition to juvenile myelomonocytic leukemia (CBL mutation-associated syndrome).2 The variant has been observed as a de novo occurrence in two unrelated individuals with features consistent with CBL-related RASopathy, with confirmed paternity in both cases.3 A minigene assay demonstrated that c.1096-1G>T results in complete skipping of exon 8, which encodes the linker region and RING finger domain critical for CBL E3 ubiquitin ligase function.4 The variant affects the RING finger domain / linker region (exons 8-9), a well-established mutational hotspot where approximately 50% of germline CBL mutations cluster and no benign variation is observed.5 The variant is extremely rare in population databases: absent from gnomAD v2.1 and gnomAD-Canada, and observed in a single heterozygous carrier in gnomAD v4.1 (1/1,590,474 alleles; AF = 0.00006%).6 This variant is classified as Pathogenic in ClinVar (Variation ID: 180815) by five independent clinical laboratories.7 Applying generic ACMG/AMP 2015 combination rules: PVS1 (very_strong) + PS2 (strong) + PM1 (moderate) + PS3 (supporting) + PM2 (supporting) + PP5 (supporting) is sufficient for a Pathogenic classification.8
CBL
Final classification
Pathogenic
CBL c.1096-1G>T · p.?
CBL
NM_005188.3:c.1096-1G>T is a canonical splice acceptor variant in CBL, disrupting the AG dinucleotide at the intron 7 / exon 8 boundary.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PS2 strong, PS3 supporting, PM1 moderate, PM2 supporting, PP5 supporting; combination = 1 very strong + 1 strong + 1 moderate + 3 supporting, which maps to Pathogenic.
Classification rationale
PVS1PS2PS3PM1PM2PP5
Pathogenic
CBL c.1096-1G>T
PVS1 + PS2 + PS3 + PM1 + PM2 + PP5
→
Pathogenic
1
pvs1_variant_assessmentspliceai ↗
2
pvs1_gene_contextPMID:25952305 ↗
8
generic_acmg_combination_rules
Gene diagram
· NM_005188.3 · variants mapped to exon structure
CBL
NM_005188.3
Fetching transcript structure from UCSC…
Applied criteria · 6 applied · 8 assessed
Applied · 6
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
This variant affects the canonical splice acceptor site of CBL intron 7 (c.1096-1G>T), disrupting the AG dinucleotide at position -1. Germline loss-of-function variants in CBL are an established disease mechanism for CBL mutation-associated syndrome (a RASopathy with predisposition to JMML). SpliceAI predicts acceptor loss with a max delta score of 0.99. Under ClinGen SVI PVS1 recommendations (PMC6185798), canonical ±1,2 splice site variants in genes with an established LoF mechanism are assigned PVS1 at full strength.
Canonical splice acceptor site disruption at intron 7 / exon 8 boundary (AG>AT)CBL germline LoF is an established disease mechanism for RASopathy (PVS1 gene gate: eligible)SpliceAI delta score 0.99 (acceptor loss)
✓
PS2
strong
Pathogenic
NM_005188.3:c.1096-1G>T has been observed as a de novo occurrence in two unrelated individuals with features consistent with CBL-related RASopathy, with confirmed paternity in both cases. Martinelli et al. (2015) reported this variant as de novo in a sporadic case (patient 827-10) with parental DNA confirmation. Seaby et al. (2017) reported the same variant as de novo in a child with atypical hemolytic uremic syndrome and RASopathy features.
De novo in case 827-10parental DNA confirmed (Martinelli 2015Table 1)
✓
PS3
supporting
Pathogenic
A minigene assay performed by Martinelli et al. (2015) demonstrated that c.1096-1G>T results in aberrant splicing with complete skipping of exon 8, which encodes essential regions of the linker region and RING finger domain. This well-established in vitro functional assay confirms a damaging effect on mRNA processing and protein function. Applied at supporting level because this evidence confirms the null effect already captured by PVS1 rather than providing independent functional evidence of a distinct mechanism.
Minigene assay in COS1 cells: c.1096-1G>T causes complete skipping of exon 8 (Martinelli 2015Figure 2B)
✓
PM1
moderate
Pathogenic
The c.1096-1G>T variant affects splicing of exon 8, which encodes the linker region and RING finger domain of CBL. Exons 8 and 9 are well-established mutational hotspots: approximately 50% of germline CBL mutations affect Tyr371 within this region, and splice site mutations affecting introns 7 and 8 account for approximately 10% of CBL defects in myeloid malignancies. The affected region is a critical functional domain with a high density of pathogenic variants and no benign variation.
Exons 8-9 encode the RING finger domain and linker region — a known mutational hotspot (Martinelli 2015)Approximately 10% of CBL defects in myeloid malignancies are splice site mutations affecting introns 7-8 (Kales et al. 2010cited in Martinelli 2015)
✓
PM2
supporting
Pathogenic
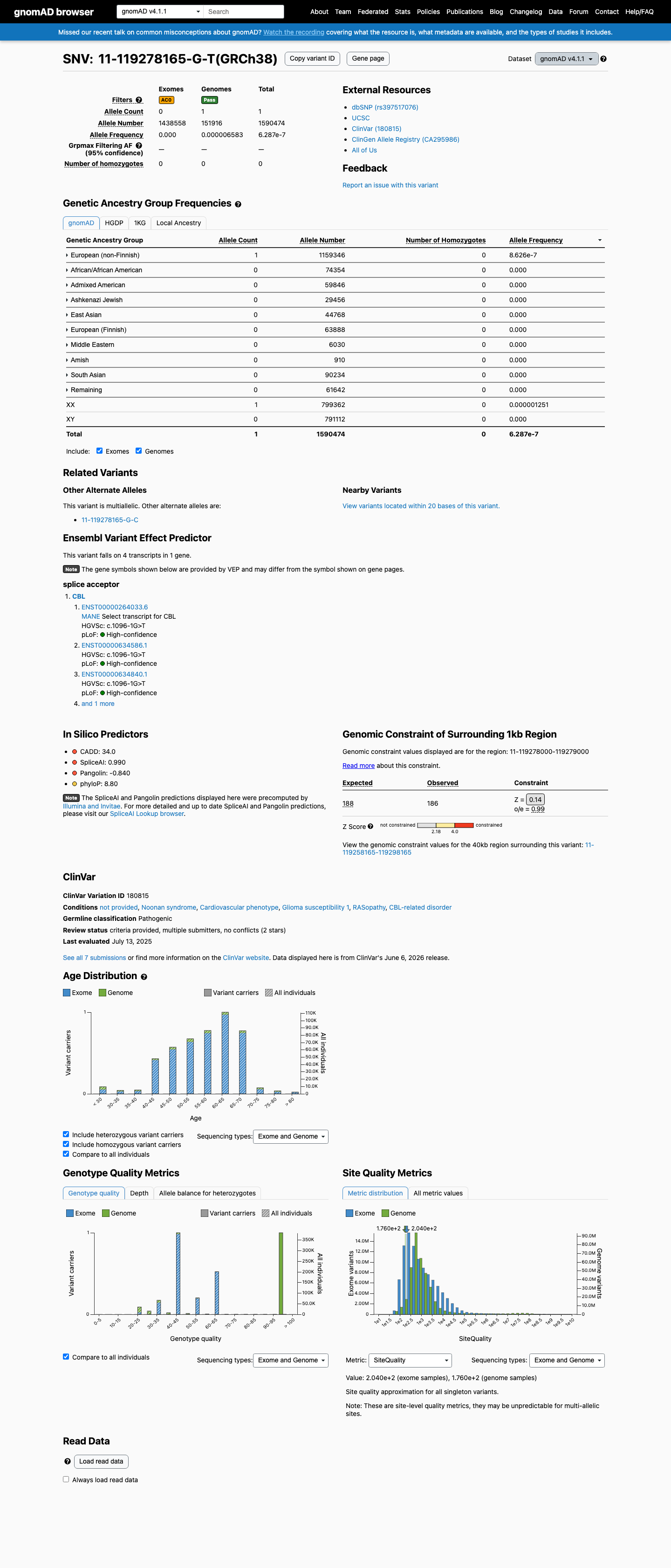
NM_005188.3:c.1096-1G>T is absent from gnomAD v2.1 and gnomAD-Canada v1.0. In gnomAD v4.1, it is observed in a single heterozygous carrier (1/1,590,474 alleles; AF = 6.29e-07, ~0.00006%), well below the PM2 threshold of 0.1%. The variant is absent from all subpopulations except European (non-Finnish) where it is present in 1/1,159,346 alleles.
Absent from gnomAD v2.1 (exomes)gnomAD v4.1: 1/1590
✓
PP5
supporting
Pathogenic
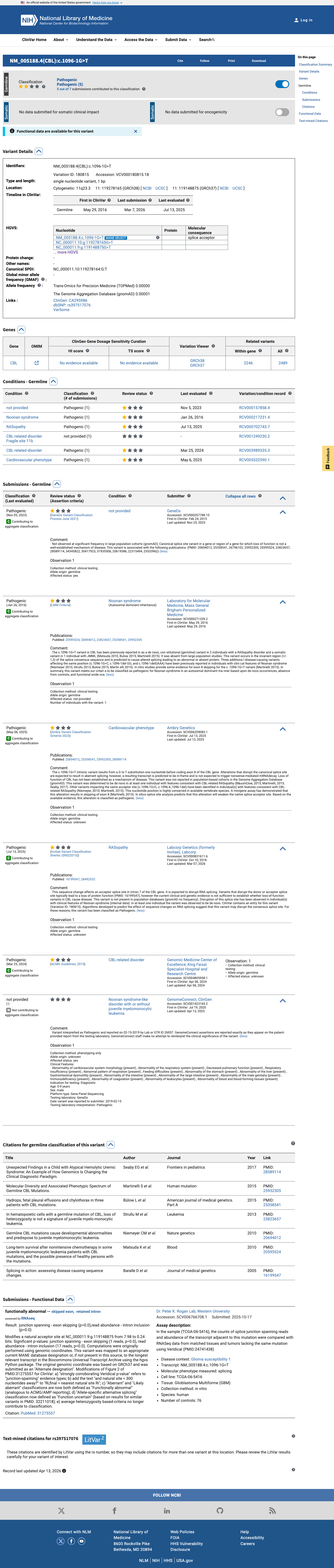
NM_005188.3:c.1096-1G>T is classified as Pathogenic in ClinVar (Variation ID: 180815) by five clinical laboratories (GeneDx, Laboratory for Molecular Medicine, Ambry Genetics, Invitae/Labcorp, and King Faisal Specialist Hospital). While the review status is 'criteria provided, single submitter' and no expert panel review is available, multiple independent clinical laboratories have reached the same conclusion based on overlapping evidence.
ClinVar Variation ID 180815: Pathogenic5 clinical laboratoriesMultiple independent submitters reaching same classification
Assessed · not applied
Pathogenic
PS4
Insufficient data to calculate a statistically significant difference in variant prevalence between affected individuals and the general population.
PP1
No segregation data are available for this variant.
PP4
CBL-related RASopathy has overlapping features with Noonan syndrome and other RASopathies; the phenotype is not sufficiently specific to a single genetic etiology to apply PP4.
Benign
BS2
The variant is observed in a single heterozygous carrier in gnomAD v4.1 (1/1,590,474 alleles).
BS3
Well-established in vitro functional studies (minigene assay, Martinelli 2015) demonstrate that c.1096-1G>T causes aberrant splicing with complete skipping of exon 8, supporting a damaging rather than benign effect.
BS4
No segregation data are available to assess for lack of cosegregation with disease in affected families.
BP4
Multiple in silico tools predict a damaging effect on splicing.
BP6
ClinVar classifies this variant as Pathogenic (Variation ID: 180815), not Benign or Likely Benign.
N/A · 12
PS1 · PM5 · PM6 · PP2 · PP3 · BA1 · BS1 · BP1 · BP2 · BP3 · BP5 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 6.28743e-07; MAF= 0.00006%, 1/1590474 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.62555e-07; MAF= 0.00009%, 1/1159346 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.3e-05%
· 1 / 1,590,474
0 hom
0 hom
European (non-Finnish) 1 / 1,159,346 |
8.6e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Pathogenic (5 clinical laboratories). (ClinVarID = 180815)
In silico
SpliceAI predicts possible splice impact for this variant (max delta score = 0.99). BayesDel score = 0.393226.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV50635311, n = 8 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 7 further PMIDs triaged but not cited — see Sources & References.
Molecular Diversity and Associated Phenotypic Spectrum of Germline CBL Mutations.
Searched
c.1096-1G>T1096-1G
Found
Reports c.1096-1G>T as a de novo heterozygous splice site variant in a boy (case 827-10) with prenatal pleural effusions/hydrops, severe feeding difficulties, failure to thrive, short stature, hypotonia, psychomotor retardation, splenomegaly, and mild pulmonary valve stenosis. Minigene assay in COS1 cells demonstrated that c.1096-1G>T causes complete skipping of exon 8, which encodes the linker region and RING finger domain. Parental DNA confirmed de novo origin.
Variant
✓ Names this variant — characterised directly
Applied to
→PVS1
met
→PS2
met
→PS3
met
→PM1
met
Why
Key paper providing de novo evidence, functional confirmation of exon 8 skipping, and clinical phenotype. Referenced in PVS1, PS2, PS3, PM1, and PP4 assessments.
The c.1096-1G>T transversion found in case 827-10 had not been previously described... Sequencing of the processed transcript RT-PCR products revealed aberrant splicing of the mutant allele, resulting in loss of the entire exon 8.
Location Results: Mutation analysis (paragraphs 1-3); Molecular characterization of splice site mutations (paragraph 2); Clinical features (paragraph 4); Figure 2B · Context Minigene assay using pSPL3 exon trapping vector, transiently transfected in COS1 cells · full text
Unexpected Findings in a Child with Atypical Hemolytic Uremic Syndrome: An Example of How Genomics Is Changing the Clinical Diagnostic Paradigm.
Searched
c.1096-1G>T1096-1G
Found
Reports a de novo heterozygous CBL c.1096-1G>T splice site variant identified by whole-genome sequencing in a child with atypical hemolytic uremic syndrome. The variant was predicted pathogenic and considered the likely explanation for RASopathy features. The paper highlights the expanding clinical spectrum of CBL-related disorders.
Variant
✓ Names this variant — characterised directly
Applied to
→PS2
met
Why
Provides a second independent de novo observation, supporting PS2 at strong level.
Filtering parameters reduced 51 variants to one heterozygous splicing variant in CBL (c.1096-1G>T) predicted to be pathogenic following application of the American College of Medical Genetics and Genomics guidelines.
Location Case presentation; Figure 1 legend · full text
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
20694012 ↗
Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia.
CLINVAR
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
25173338 ↗
2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC).
CLINVAR