NM_001127511.3:c.793C>T (p.Arg265Ter) is a nonsense variant in exon 7 of APC, a gene where loss of function is the established disease mechanism for familial adenomatous polyposis.1 The variant meets PVS1 at Very Strong strength: null variant in a LOF-established gene, expected to trigger nonsense-mediated decay (InSiGHT VCEP v2.1.0 modified decision tree).2 The variant has been observed in multiple unrelated families with classic FAP: a 3-generation Malaysian Chinese family (PMID:12901799) and 3 affected individuals from 2 Brazilian families (PMID:30897307), meeting PS4 at Moderate strength (estimated 3.5 phenotype points).3 The variant is absent from gnomAD v2.1.1 and present at extremely low frequency in gnomAD v4.1 (AF=2.48e-6, grpmax FAF=8e-7), meeting PM2 at Supporting strength under APC VCEP population thresholds.4 Under the APC VCEP v2.1.0 combination rules (Rule 22): one Very Strong criterion (PVS1) + one Moderate criterion (PS4) + one Supporting criterion (PM2) reaches Pathogenic classification. The ClinGen InSiGHT expert panel independently classified this variant as Pathogenic (ClinVar variation 184999).5
APC
Final classification
Likely Pathogenic
APC c.793C>T · p.Arg265Ter
APC
NM_001127511.3:c.793C>T (p.Arg265Ter) is a nonsense variant in exon 7 of APC, a gene where loss of function is the established disease mechanism for familial adenomatous polyposis.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for APC Version 2.1.0 v2.1.0 criteria-combination framework: matched Rule18 (1 Pathogenic.Very Strong + 1 Pathogenic.Moderate) with applied criteria: PVS1 very strong, PS4 moderate, PM2 supporting, PP5 supporting; maps to Likely Pathogenic.
Classification rationale
PVS1PS4PM2PP5
Likely Pathogenic
APC c.793C>T
PVS1 + PS4 + PM2 + PP5
→
Likely Pathogenic
Gene diagram
· NM_001127511.3 · variants mapped to exon structure
APC
NM_001127511.3
Fetching transcript structure from UCSC…
Applied criteria · 4 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
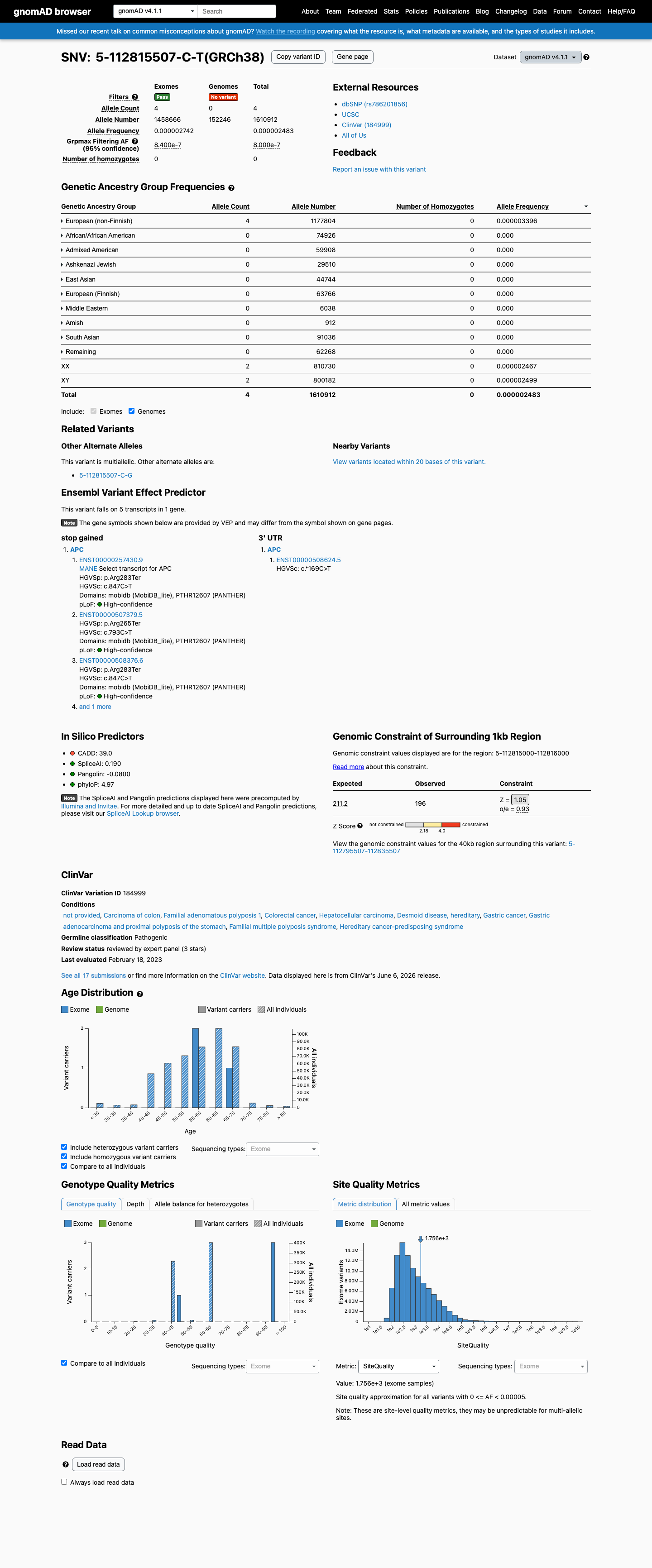
This variant is present in gnomAD v4.1 (AF= 2.48307e-06; MAF= 0.00025%, 4/1610912 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.39615e-06; MAF= 0.00034%, 4/1177804 alleles, homozygotes = 0); grpmax FAF= 8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,610,912
0 hom · FAF 8e-05%
0 hom · FAF 8e-05%
European (non-Finnish) 4 / 1,177,804 |
0.00034% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

In silico
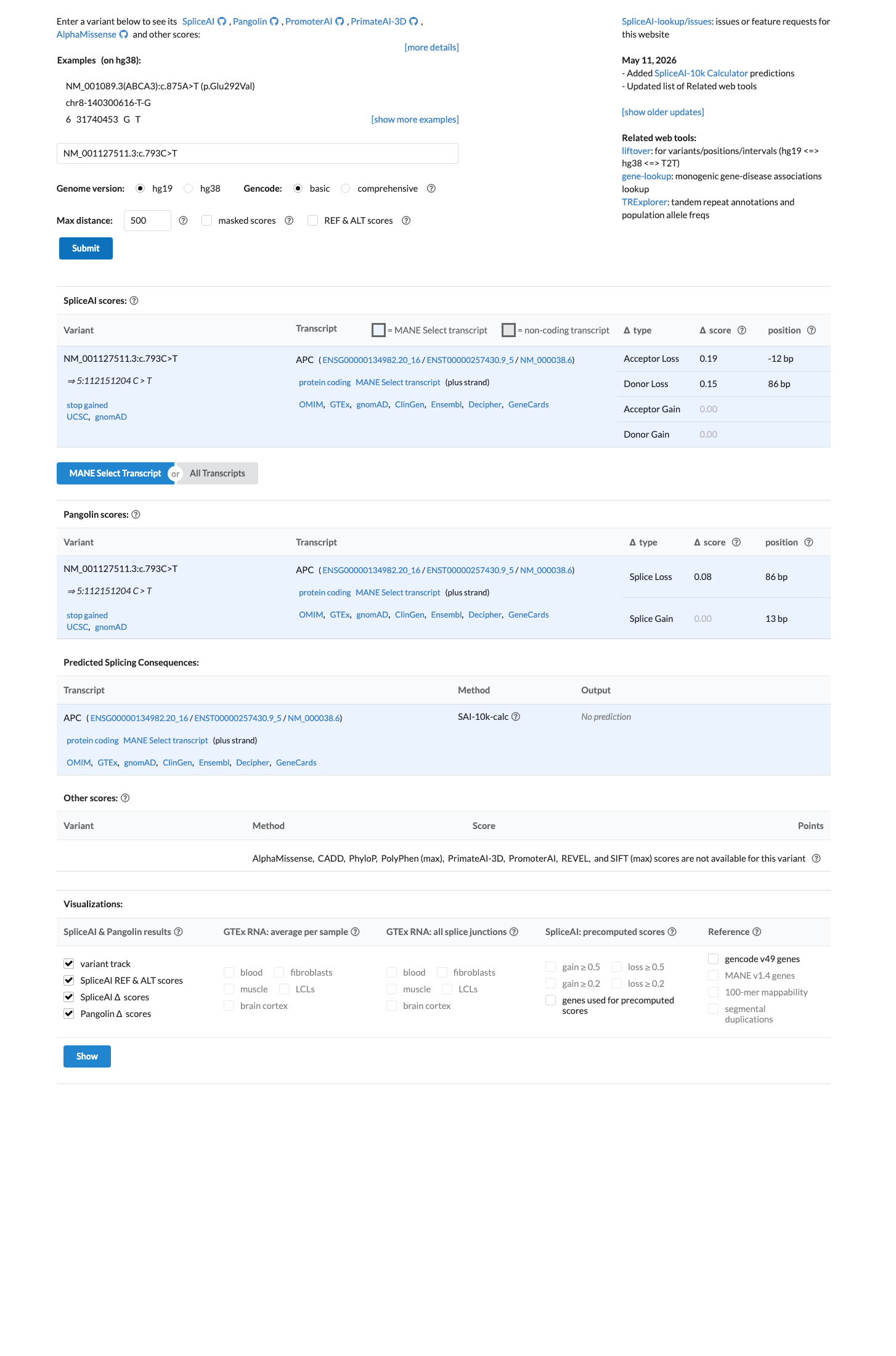
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.19). BayesDel score = 0.66.
Functional
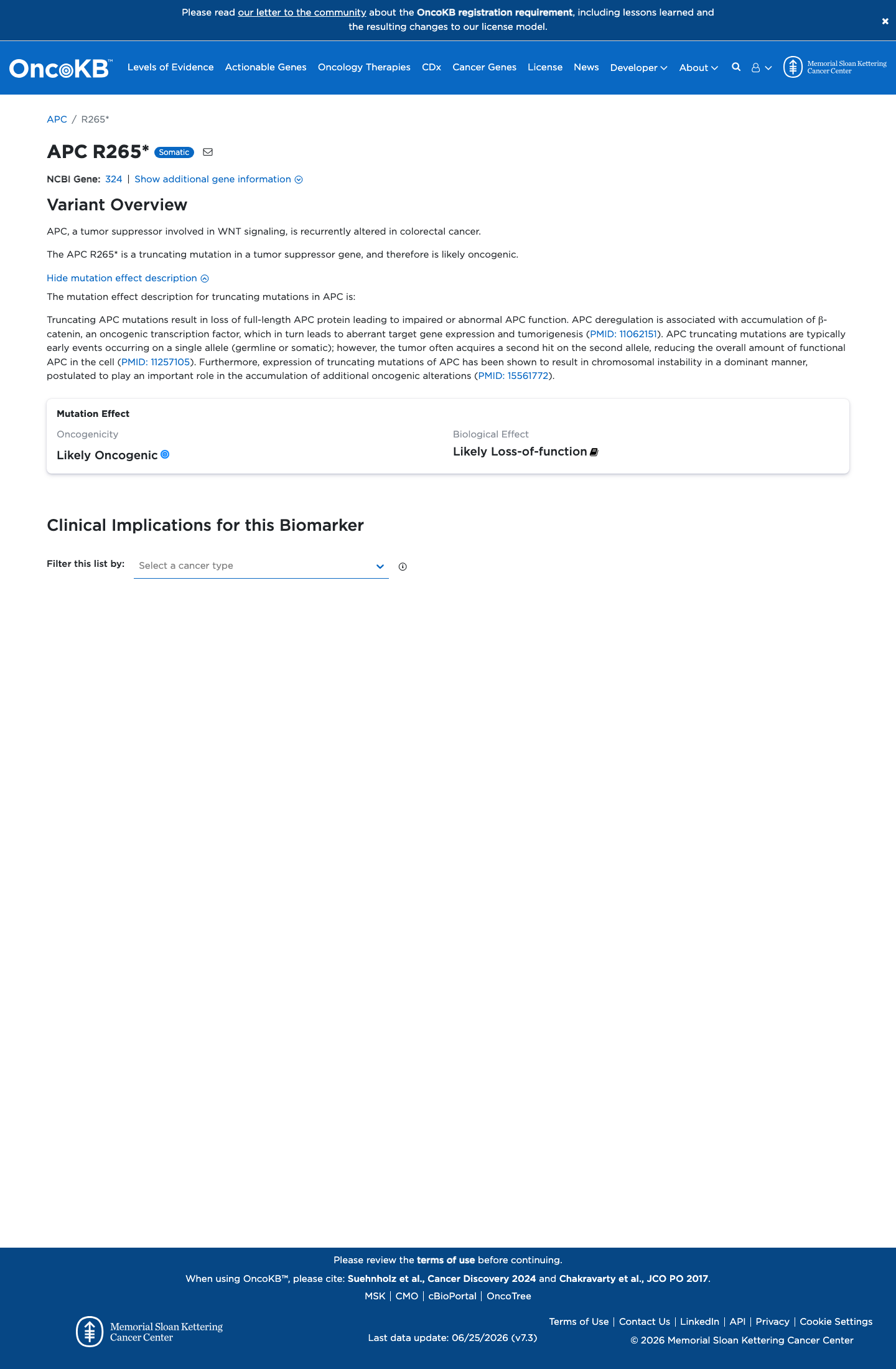
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
A nonsense mutation in exon 8 of the APC gene (Arg283Ter) causes clinically variable FAP in a Malaysian Chinese family.
Searched
c.847C>TArg283TerR283*C847Tnonsense mutation exon 8
Found
A nonsense mutation in exon 8 of APC (NM_000038.6:c.847C>T, p.Arg283Ter) — identical to NM_001127511.3:c.793C>T under alternate transcript nomenclature — was identified as causative in a 3-generation Malaysian Chinese family with FAP. The variant segregated with disease in affected family members. SSCP analysis confirmed heterozygous C>T transition. Computational analysis predicted possible disruption of splicing enhancer motifs.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
Why
Variant-specific segregation and phenotype data confirmed; referenced in PS4 (Moderate) and PP1 (not met — insufficient documented meioses).
Sequence analysis revealed that the affected individuals are heterozygous for a C847T transition, whilst all the unaffected family members and control individuals are homozygous C at the same position. This nucleotide substitution generates a stop codon at amino acid position 283, in place of the usual arginine (Arg283Ter).
Location Abstract; Results paragraph describing sequencing confirmation · full text
Genotype-phenotype correlation in 99 familial adenomatous polyposis patients: A prospective prevention protocol.
Searched
c.847C>TArg283TerR283*codon 283847
Found
In a Brazilian cohort of 99 FAP patients from 35 families, the variant NM_000038.6:c.847C>T (p.Arg283Ter) — identical to NM_001127511.3:c.793C>T — was identified in 3 individuals from 2 unrelated families (families 10 and 21). All affected individuals exhibited classic FAP; one individual had osteoma. The variant was among 26 different APC pathogenic variants characterized in the study.
Variant
✓ Names this variant — characterised directly
Applied to
→PS4 supports · met
→PVS1 supports · met
Why
Variant-specific phenotype data in multiple unrelated families confirmed; referenced in PS4 (Moderate) and PVS1 assessments.
p.Arg283Ter(c.847C>T) 1 (Family 10) ... p.Arg283Ter(c.847C>T) 2 (Family 21)
Location Table 1 (families 10 and 21); Table 7 (codon 283 phenotypic correlations) · Context Prospective single-center cohort study; clinical phenotyping by colonoscopy, UGIE, CT, MRI, Doppler ultrasound, ophthalmoscopy · full text
Sources & reference links
9Sources
Triaged references · 6 PMIDs not cited in assessment
11062151 ↗
Expression of beta-catenin and full-length APC protein in normal and neoplastic colonic tissues.
ONCOKB
15561772 ↗
Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability.
ONCOKB
1338764 ↗
Screening for germ-line mutations in familial adenomatous polyposis patients: 61 new patients and a summary of 150 unrelated patients.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR