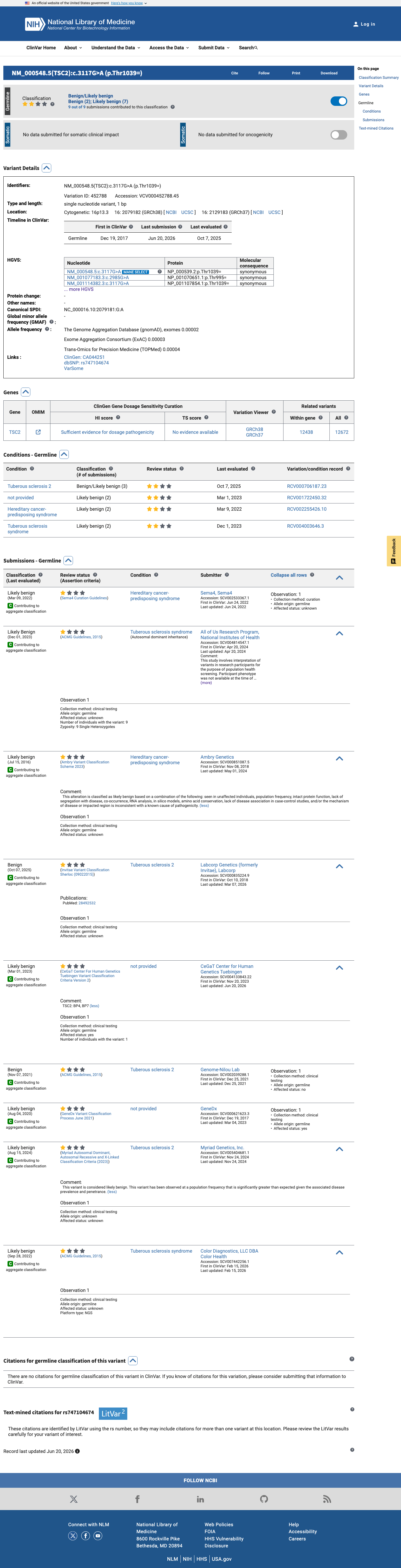
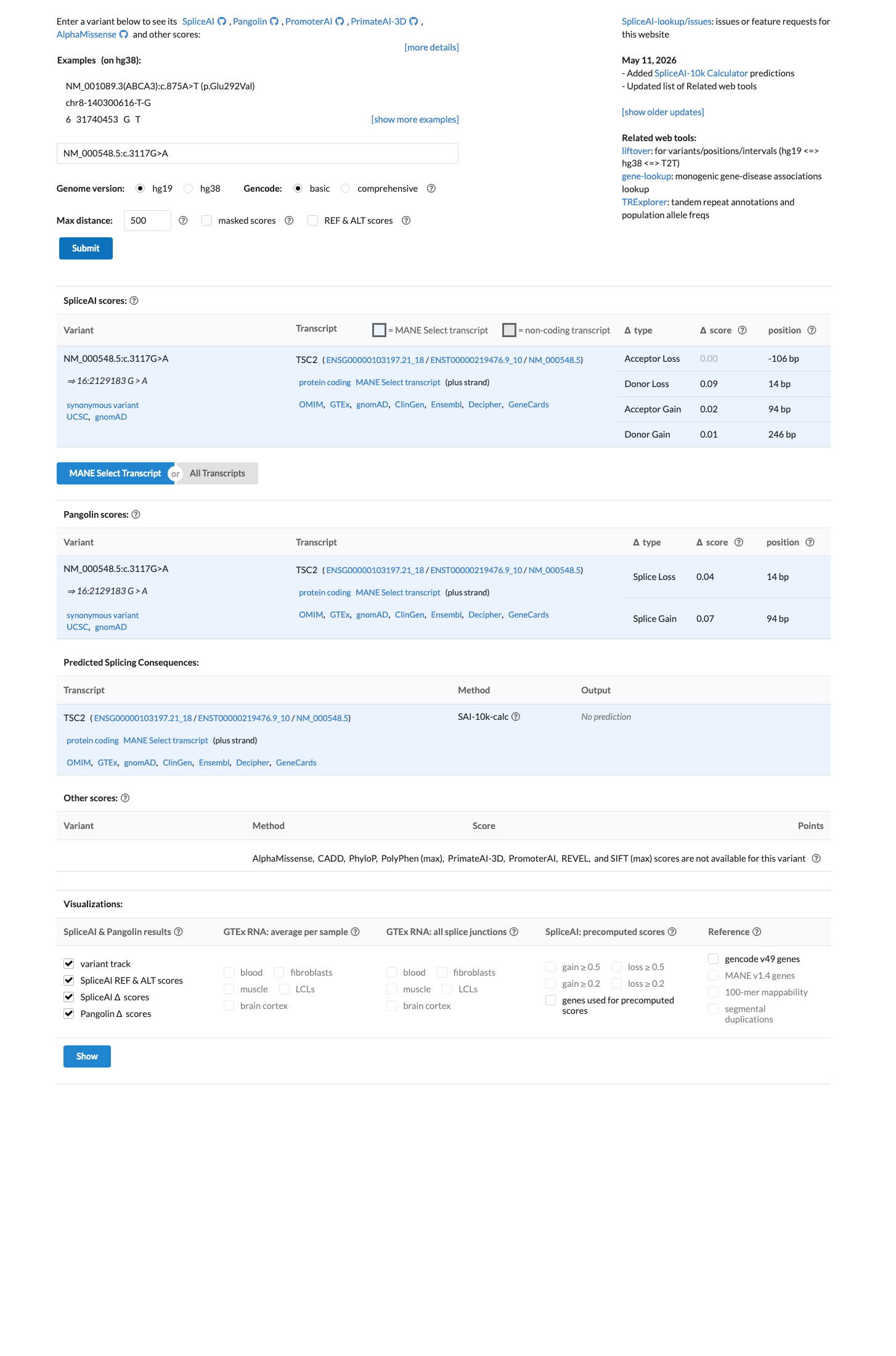
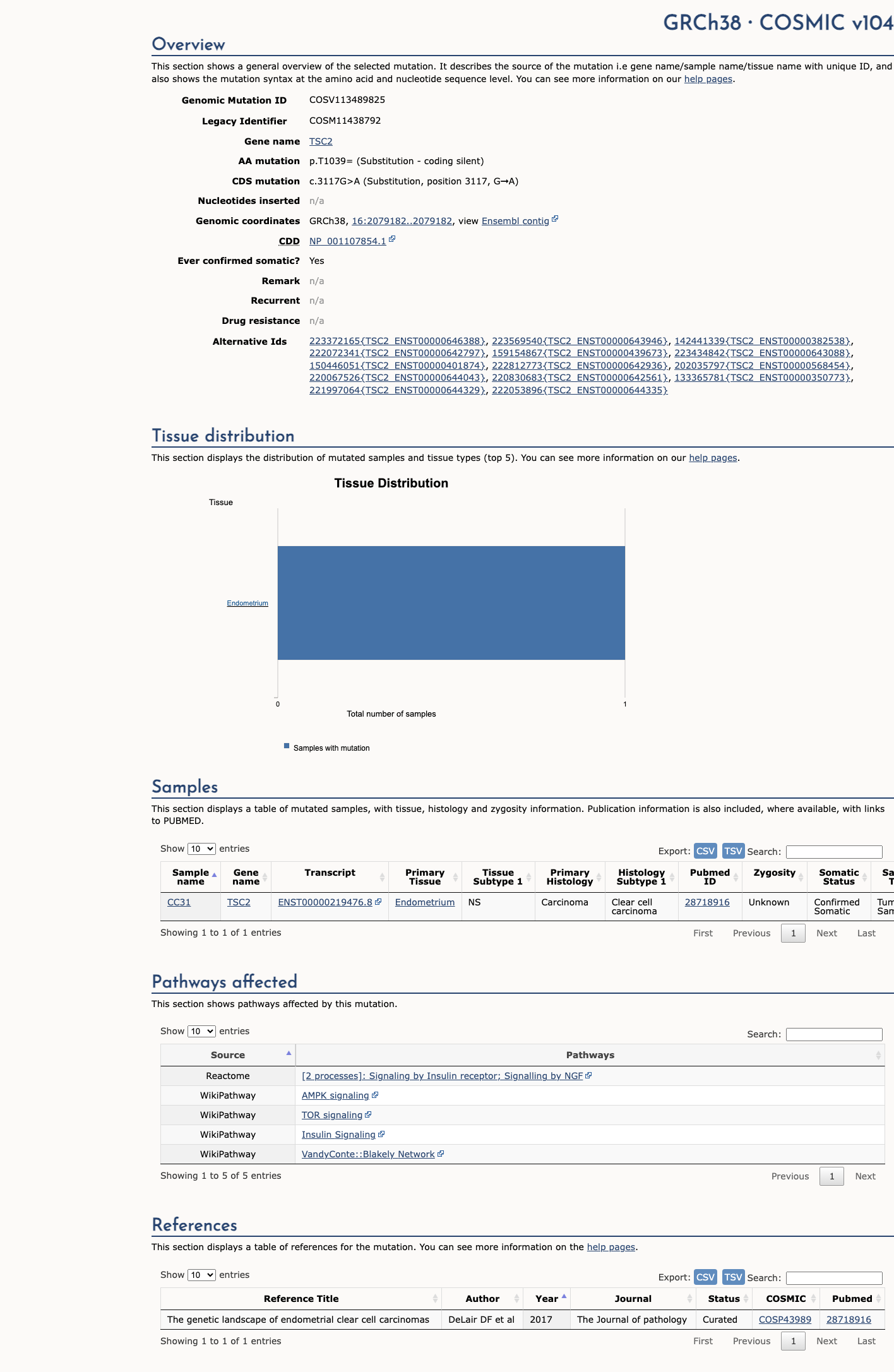
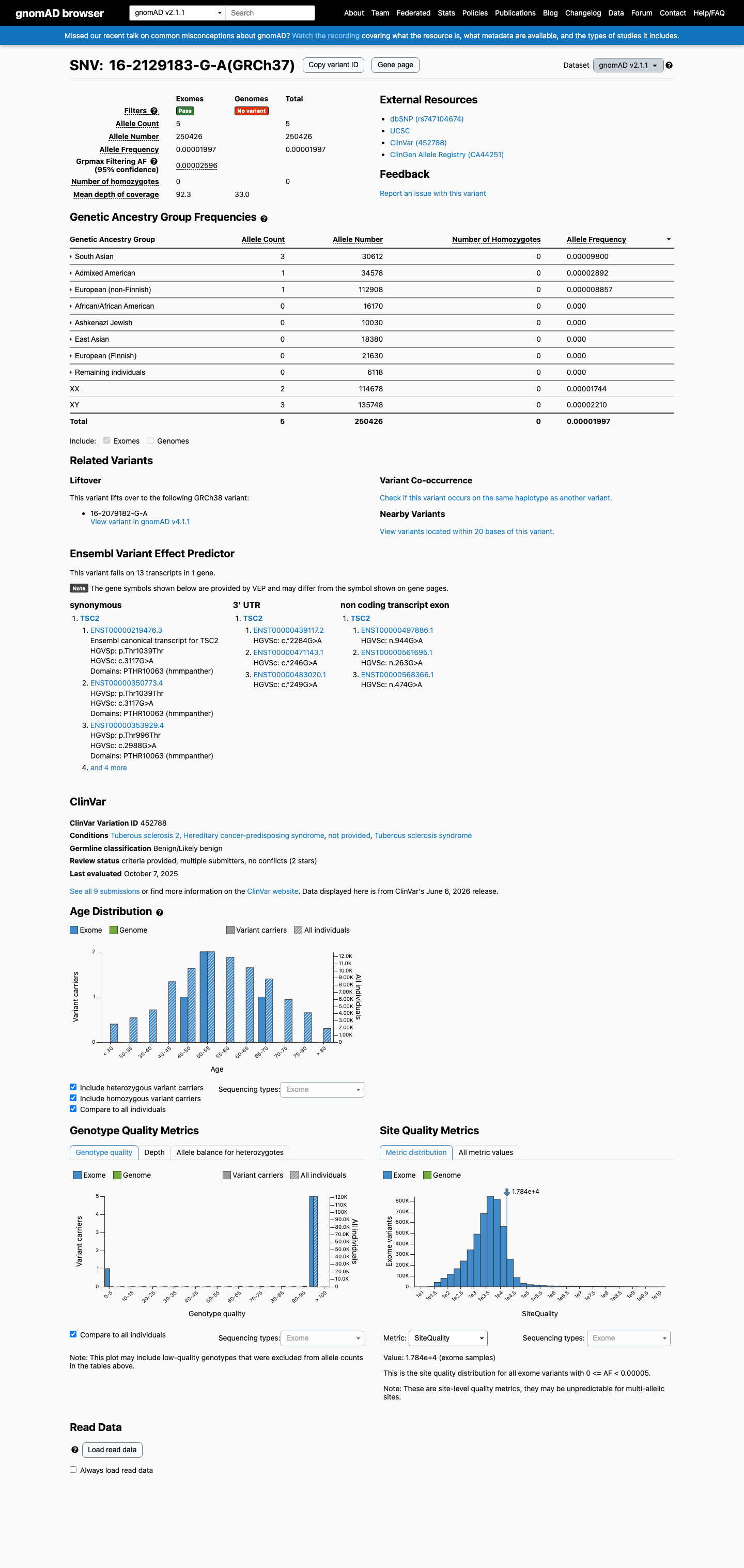
NM_000548.5:c.3117G>A is a synonymous variant in TSC2. This variant does not alter the amino acid sequence (p.Thr1039=). SpliceAI predicts no significant splice impact (max delta 0.09). The variant is absent from gnomAD v2.1. In gnomAD v4.1, it is observed at extremely low frequency (AF=0.00155%, 25/1,612,750 alleles, 0 homozygotes) across multiple ancestry groups, with highest subpopulation frequency in Admixed Americans (AF=0.00667%). No statistically significant mutational hotspot or well-characterized functional domain has been identified. OncoKB reports Unknown Oncogenic Effect with no curated literature specific to this variant. COSMIC reports 1 somatic occurrence (COSV113489825). ClinVar reports this variant as Likely benign by 5 clinical laboratories, Benign by 2, and Likely Benign by 1, with criteria provided by single submitters and no expert panel review.1
NM_000548.5:c.3117G>A is a synonymous variant in TSC2. This variant does not alter the amino acid sequence (p.Thr1039=). SpliceAI predicts no significant splice impact (max delta 0.09). The variant is absent from gnomAD v2.1. In gnomAD v4.1, it is observed at extremely low frequency (AF=0.00155%, 25/1,612,750 alleles, 0 homozygotes) across multiple ancestry groups, with highest subpopulation frequency in Admixed Americans (AF=0.00667%). No statistically significant mutational hotspot or well-characterized functional domain has been identified. OncoKB reports Unknown Oncogenic Effect with no curated literature specific to this variant. COSMIC reports 1 somatic occurrence (COSV113489825). ClinVar reports this variant as Likely benign by 5 clinical laboratories, Benign by 2, and Likely Benign by 1, with criteria provided by single submitters and no expert panel review.
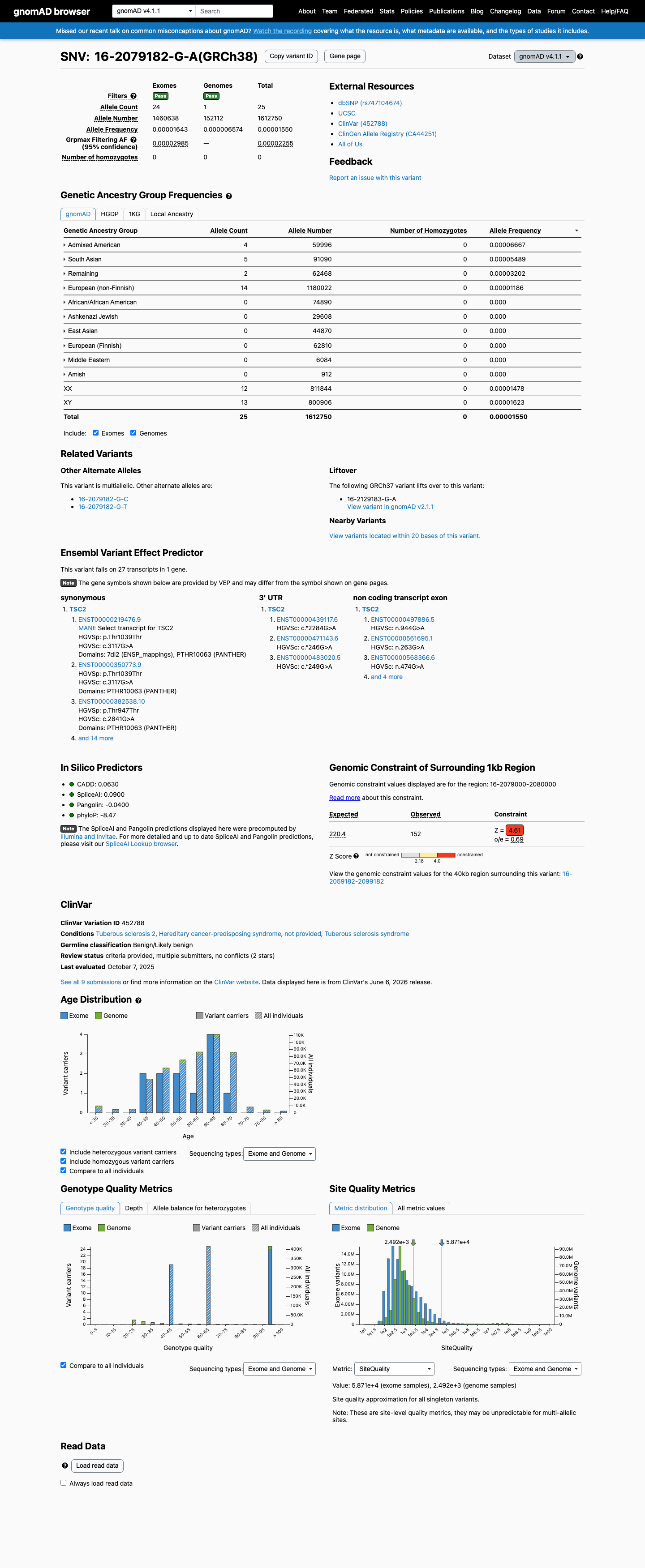
0 hom · FAF 0.0023%
Admixed American 4 / 59,996 |
0.0067% |
South Asian 5 / 91,090 |
0.0055% |
Remaining individuals 2 / 62,468 |
0.0032% |
European (non-Finnish) 14 / 1,180,022 |
0.0012% |
0 hom
0 hom