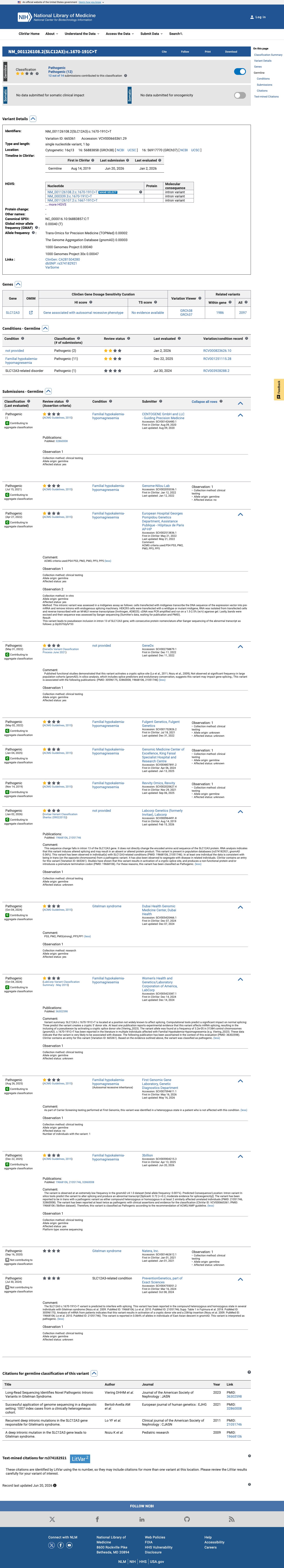
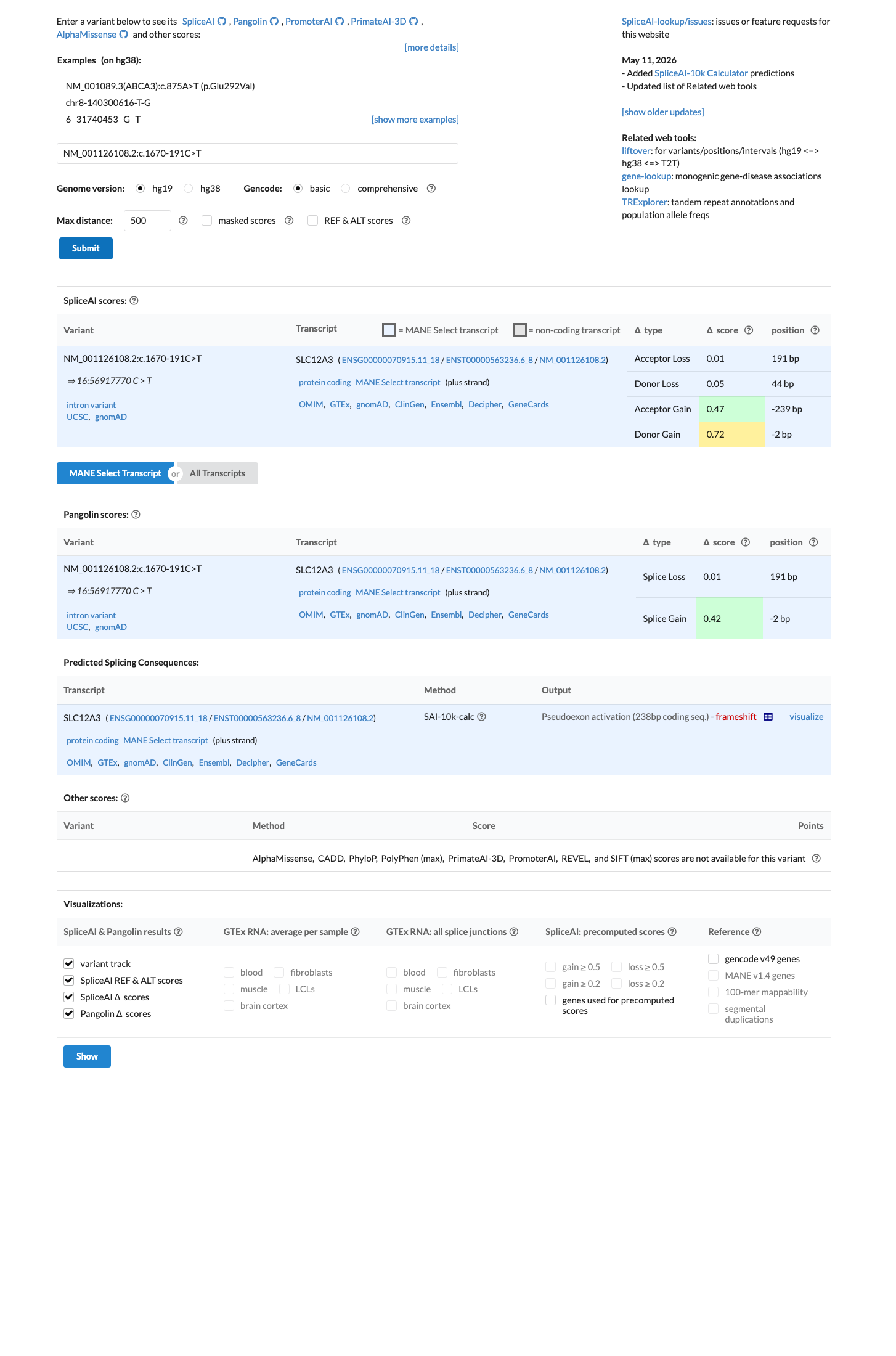
Functional studies from three independent research groups demonstrate that c.1670-191C>T creates a novel splice donor site in intron 13 of SLC12A3, resulting in 238bp cryptic exon inclusion with a premature termination codon and loss of functional NCC protein expression. The variant is extremely rare in population databases, with a maximum allele frequency of 0.064% in East Asians (gnomAD v2.1) and no homozygotes observed across all datasets, meeting the PM2 threshold.1 SpliceAI predicts a splice-altering effect with a max delta score of 0.72 (donor gain), consistent with the experimentally observed creation of a novel splice donor site.2 Classified as Pathogenic by 13 clinical diagnostic laboratories in ClinVar (Variation ID 665361), including Labcorp/Invitae, GeneDx, CENTOGENE, and Fulgent Genetics, with no conflicting interpretations.3 Based on generic ACMG/AMP 2015 classification rules, the combination of 1 strong (PS3), 1 moderate (PM2), and 2 supporting (PP3, PP5) criteria supports a classification of Likely Pathogenic.4
SLC12A3
Final classification
VUS
SLC12A3 c.1670-191C>T · p.?
SLC12A3
Functional studies from three independent research groups demonstrate that c.1670-191C>T creates a novel splice donor site in intron 13 of SLC12A3, resulting in 238bp cryptic exon inclusion with a premature termination codon and loss of functional NCC protein expression.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 moderate, PP3 supporting, PP5 supporting; combination = 1 moderate + 2 supporting, which maps to VUS.
Classification rationale
PM2PP3PP5
VUS
SLC12A3 c.1670-191C>T
PM2 + PP3 + PP5
→
VUS
Gene diagram
· NM_001126108.2 · variants mapped to exon structure
SLC12A3
NM_001126108.2
Fetching transcript structure from UCSC…
Applied criteria · 3 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
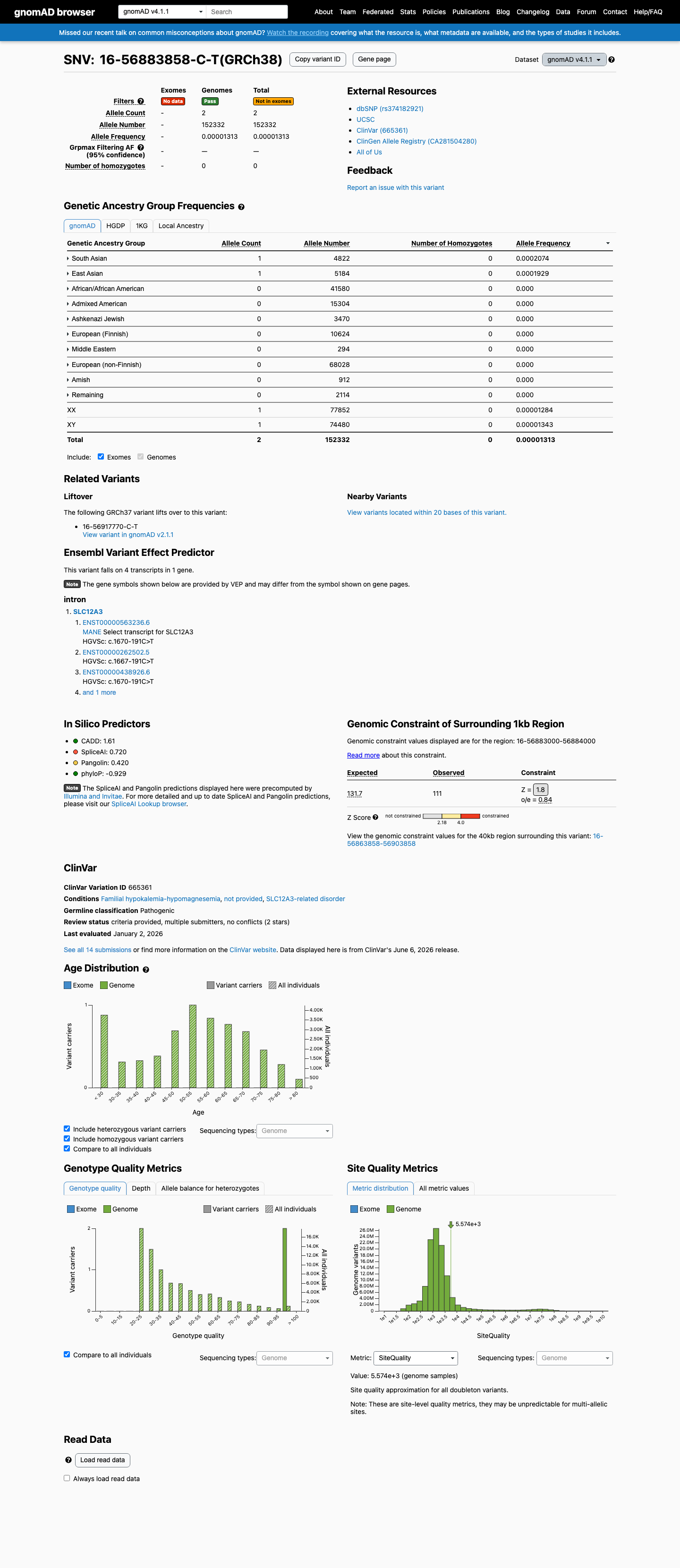
This variant is present in gnomAD v4.1 (AF= 1.31292e-05; MAF= 0.00131%, 2/152332 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.000207383; MAF= 0.02074%, 1/4822 alleles, homozygotes = 0).
v2.1
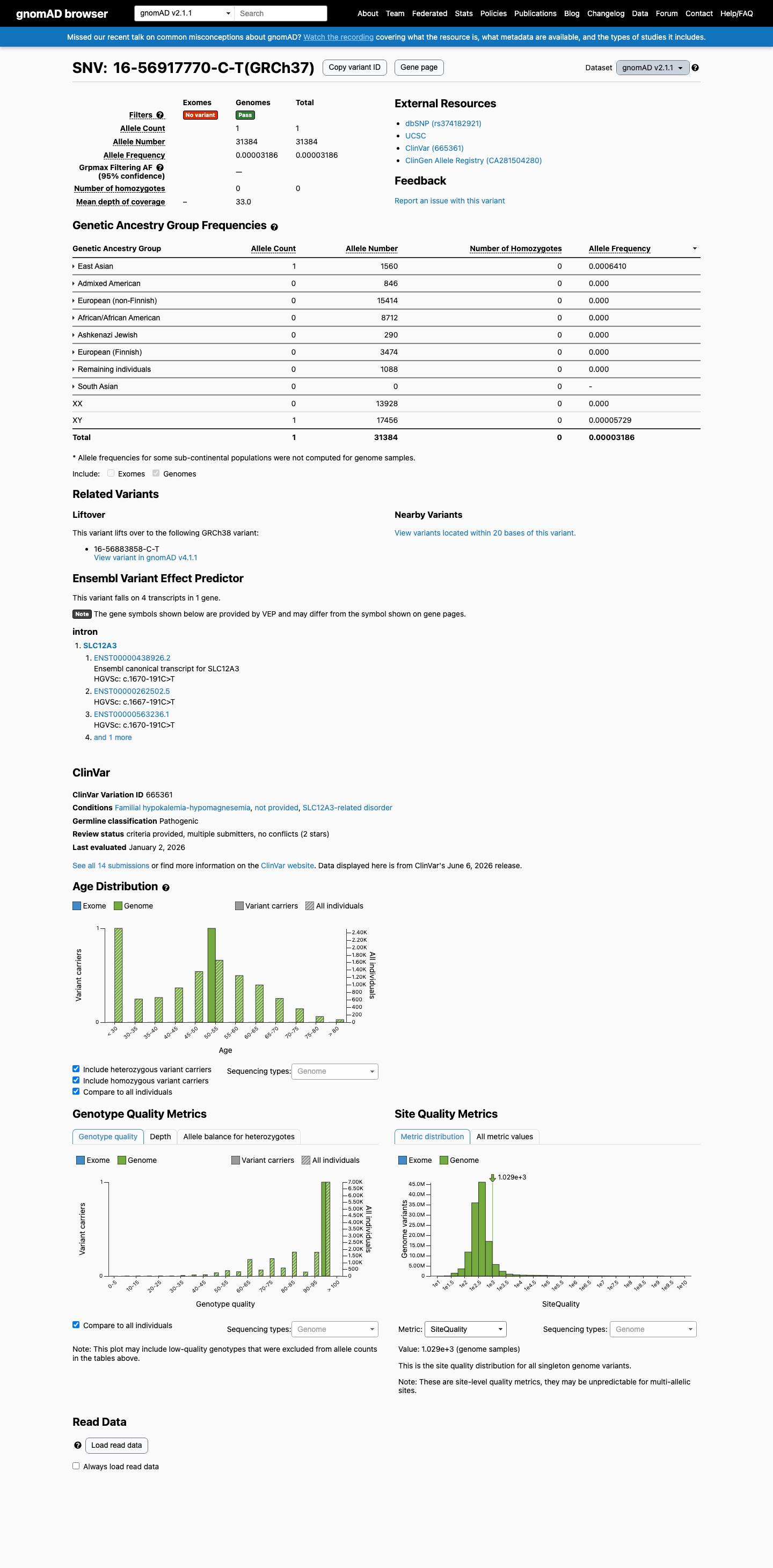
This variant is present in gnomAD v2.1 (AF= 3.18634e-05; MAF= 0.00319%, 1/31384 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.000641026; MAF= 0.06410%, 1/1560 alleles, homozygotes = 0).
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00016291951775822744, 3/18414 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0013%
· 2 / 152,332
0 hom
0 hom
South Asian 1 / 4,822 |
0.021% |
East Asian 1 / 5,184 |
0.019% |
+ 8 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
0.0032%
· 1 / 31,384
0 hom
0 hom
East Asian 1 / 1,560 |
0.064% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
0.016%
· 3 / 18,414
0 hom · FAF 0.027%
0 hom · FAF 0.027%
East Asian 2 / 1,336 |
0.15% |
European (non-Finnish) 1 / 11,738 |
0.0085% |
+ 7 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
This variant has been reported in ClinVar as Pathogenic (13 clinical laboratories). (ClinVarID = 665361)
In silico
SpliceAI predicts possible splice impact for this variant (max delta score = 0.72).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
21051746 ↗
Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
36302598 ↗
Long-Read Sequencing Identifies Novel Pathogenic Intronic Variants in Gitelman Syndrome.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
32860008 ↗
Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort.
CLINVAR