NM_033632.3:c.1697G>T (p.Trp566Leu) is a missense variant in exon 11 of FBXW7, affecting the WD40 repeat domain critical for substrate recognition. The variant is completely absent from all population databases including gnomAD v2.1, v4.1, and gnomAD-Canada (PM2_Supporting).1 FBXW7 germline loss-of-function and missense variants are established causes of an autosomal dominant neurodevelopmental syndrome with Wilms tumor predisposition (PMID:35395208). The variant alters a highly conserved tryptophan residue within the WD40 domain where pathogenic missense variants cluster (PMID:42111496), though no formal VCEP domain specification exists. The variant has been reported twice in somatic cancers (COSMIC COSV99662097), consistent with a role in tumorigenesis.2 REVEL predicts a damaging score of 0.863, though BayesDel (0.424) does not reach the damaging threshold, and no variant-specific functional studies were identified. Computational evidence alone is insufficient to meet PP3.3 No ClinVar entries, no published variant-specific functional studies, no segregation or de novo data, and no case-control evidence exist for this variant. The variant remains unclassified in all major clinical databases.4
FBXW7
Final classification
VUS
FBXW7 c.1697G>T · p.Trp566Leu
FBXW7
NM_033632.3:c.1697G>T (p.Trp566Leu) is a missense variant in exon 11 of FBXW7, affecting the WD40 repeat domain critical for substrate recognition. The variant is completely absent from all population databases including gnomAD v2.1, v4.1, and gnomAD-Canada (PM2_Supporting).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting; combination = 1 supporting, which maps to VUS.
Classification rationale
PM2
VUS
FBXW7 c.1697G>T
PM2
→
VUS
2
pvs1_gene_context
3
revelbayesdelspliceai ↗
Gene diagram
· NM_033632.3 · variants mapped to exon structure
FBXW7
NM_033632.3
Fetching transcript structure from UCSC…
Applied criteria · 1 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
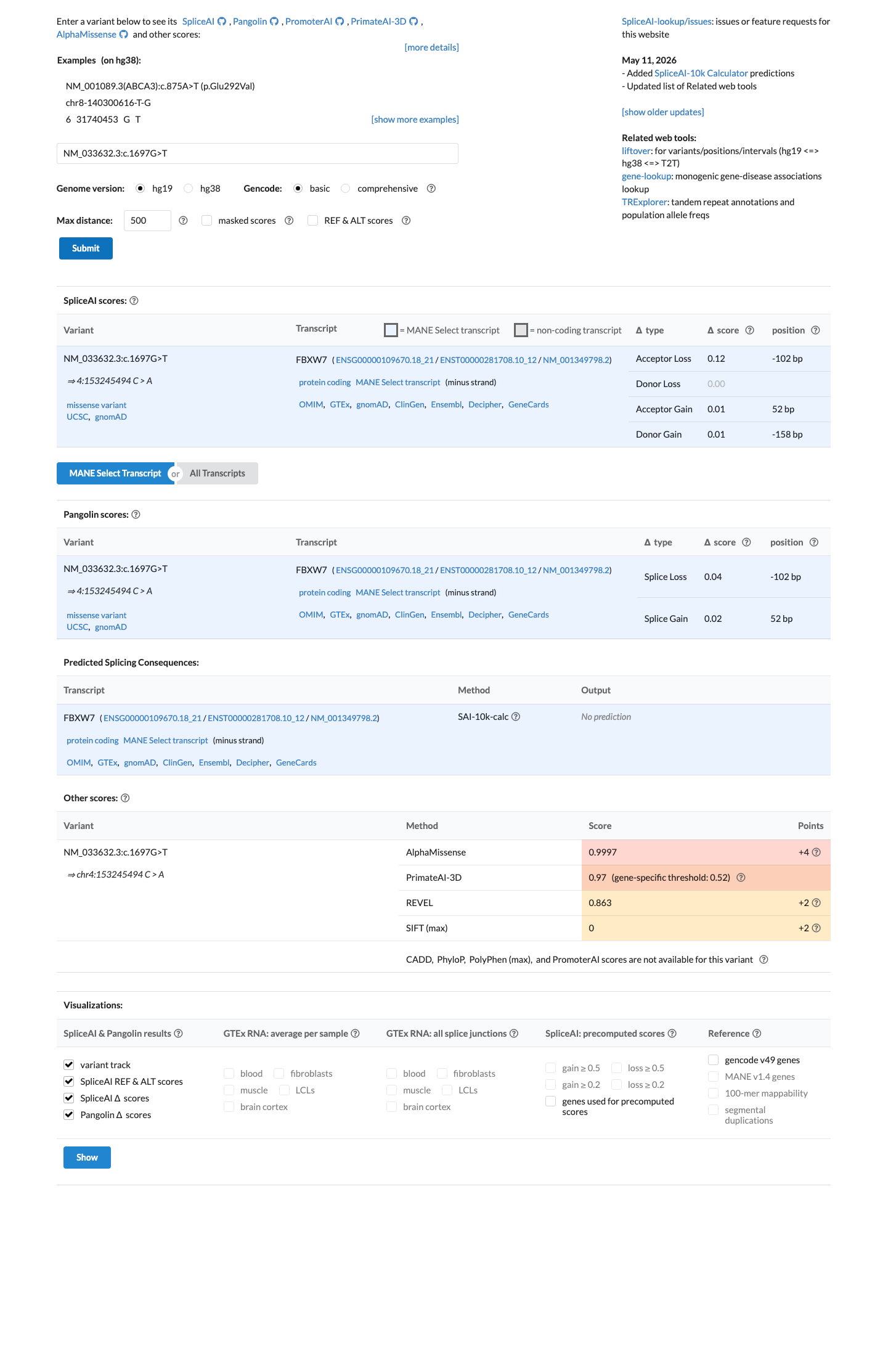
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.12). REVEL score = 0.863. BayesDel score = 0.424003.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. FBXW7, a tumor suppressor involved in protein degradation, is inactivated by mutation in various cancer types, most frequently in endometrial and colo
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99662097, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links