NM_015338.5:c.2694G>A (p.Trp898Ter) is a nonsense variant in exon 13 of 13 in ASXL1, a gene for which loss of function is an established disease mechanism.1 Under ClinGen SVI PVS1 recommendations (PMC6185798), the variant is located in the terminal exon and is not expected to undergo nonsense-mediated decay; PVS1 is applied at moderate strength.2 The variant is extremely rare in population databases with an allele frequency of 3.98 × 10⁻⁶ in gnomAD v2.1 and 1.24 × 10⁻⁶ in gnomAD v4.1, meeting PM2 at supporting level.3 No additional pathogenic or benign criteria were met; the variant is absent from ClinVar, no variant-specific functional studies exist, and no segregation or de novo data are available.4 Based on generic ACMG/AMP 2015 combination rules, one moderate criterion (PVS1_Moderate) and one supporting criterion (PM2_Supporting) are met, yielding a classification of Likely Pathogenic.5
ASXL1
Final classification
VUS
ASXL1 c.2694G>A · p.Trp898Ter
ASXL1
NM_015338.5:c.2694G>A (p.Trp898Ter) is a nonsense variant in exon 13 of 13 in ASXL1, a gene for which loss of function is an established disease mechanism.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 moderate, PM2 supporting; combination = 1 moderate + 1 supporting, which maps to VUS.
Classification rationale
PVS1PM2
VUS
ASXL1 c.2694G>A
PVS1 + PM2
→
VUS
1
pvs1_generic_framework ↗pvs1_gene_context
2
pvs1_generic_framework ↗pvs1_variant_assessment
5
generic_acmg_combination_rules
Gene diagram
· NM_015338.5 · variants mapped to exon structure
ASXL1
NM_015338.5
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
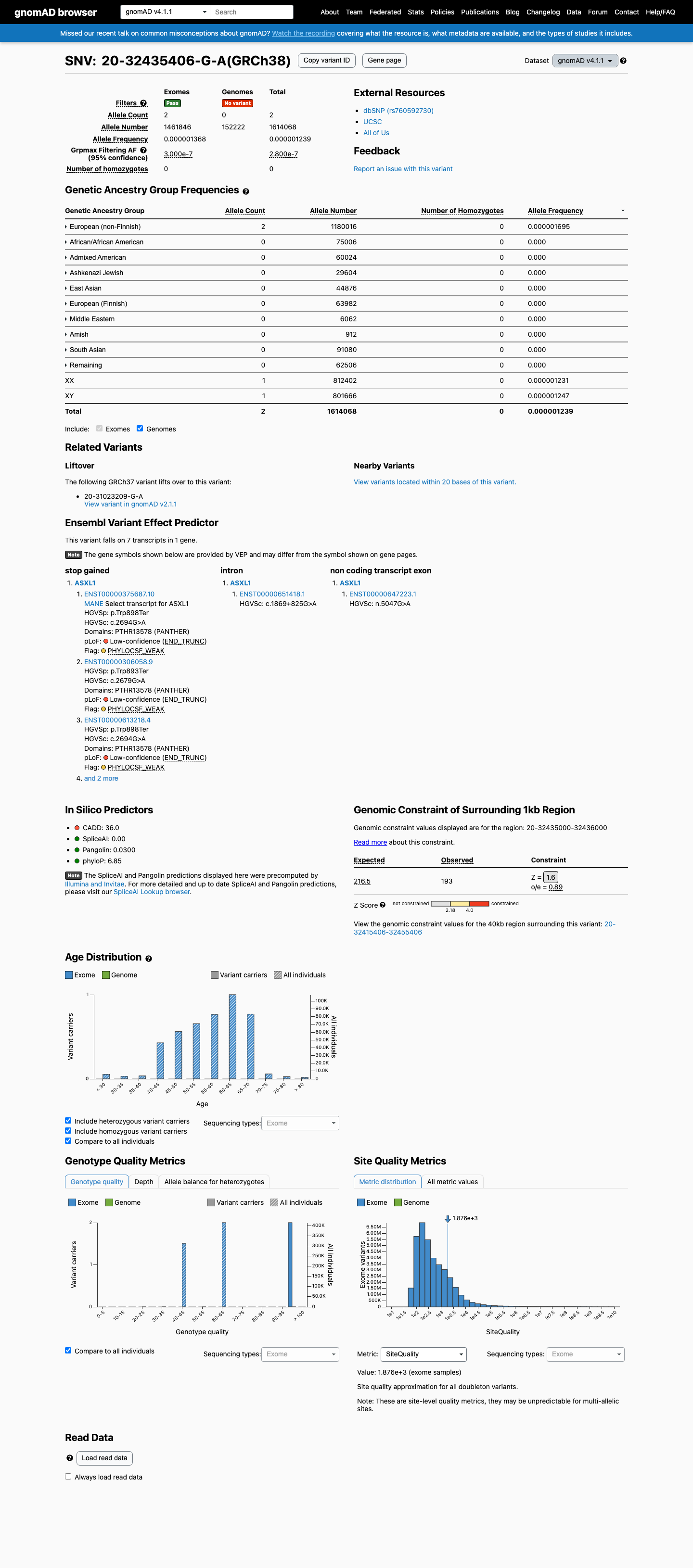
This variant is present in gnomAD v4.1 (AF= 1.23911e-06; MAF= 0.00012%, 2/1614068 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.69489e-06; MAF= 0.00017%, 2/1180016 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
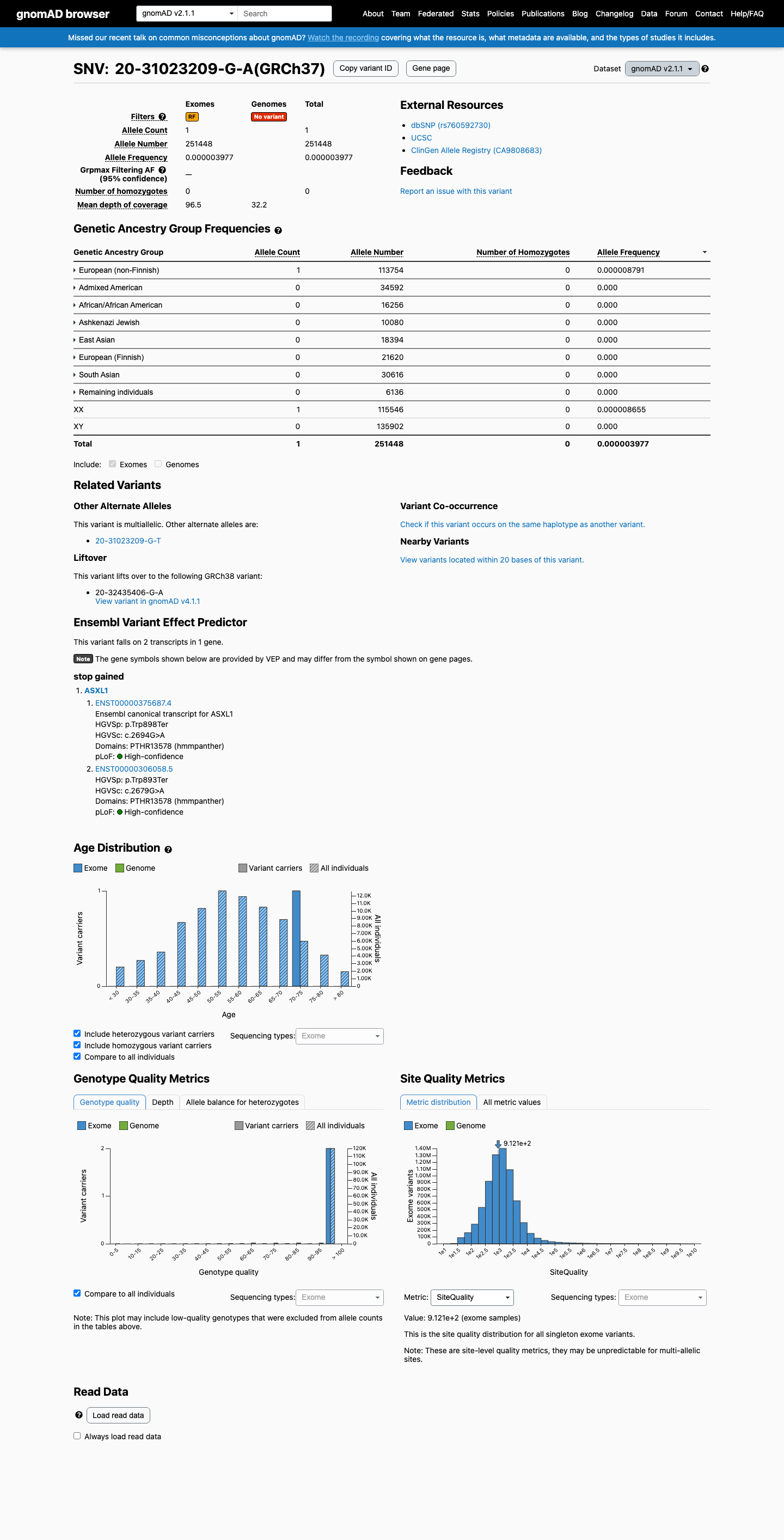
This variant is present in gnomAD v2.1 (AF= 3.97697e-06; MAF= 0.00040%, 1/251448 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.7909e-06; MAF= 0.00088%, 1/113754 alleles, homozygotes = 0).
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00012%
· 2 / 1,614,068
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
European (non-Finnish) 2 / 1,180,016 |
0.00017% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 251,448
0 hom
0 hom
European (non-Finnish) 1 / 113,754 |
0.00088% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
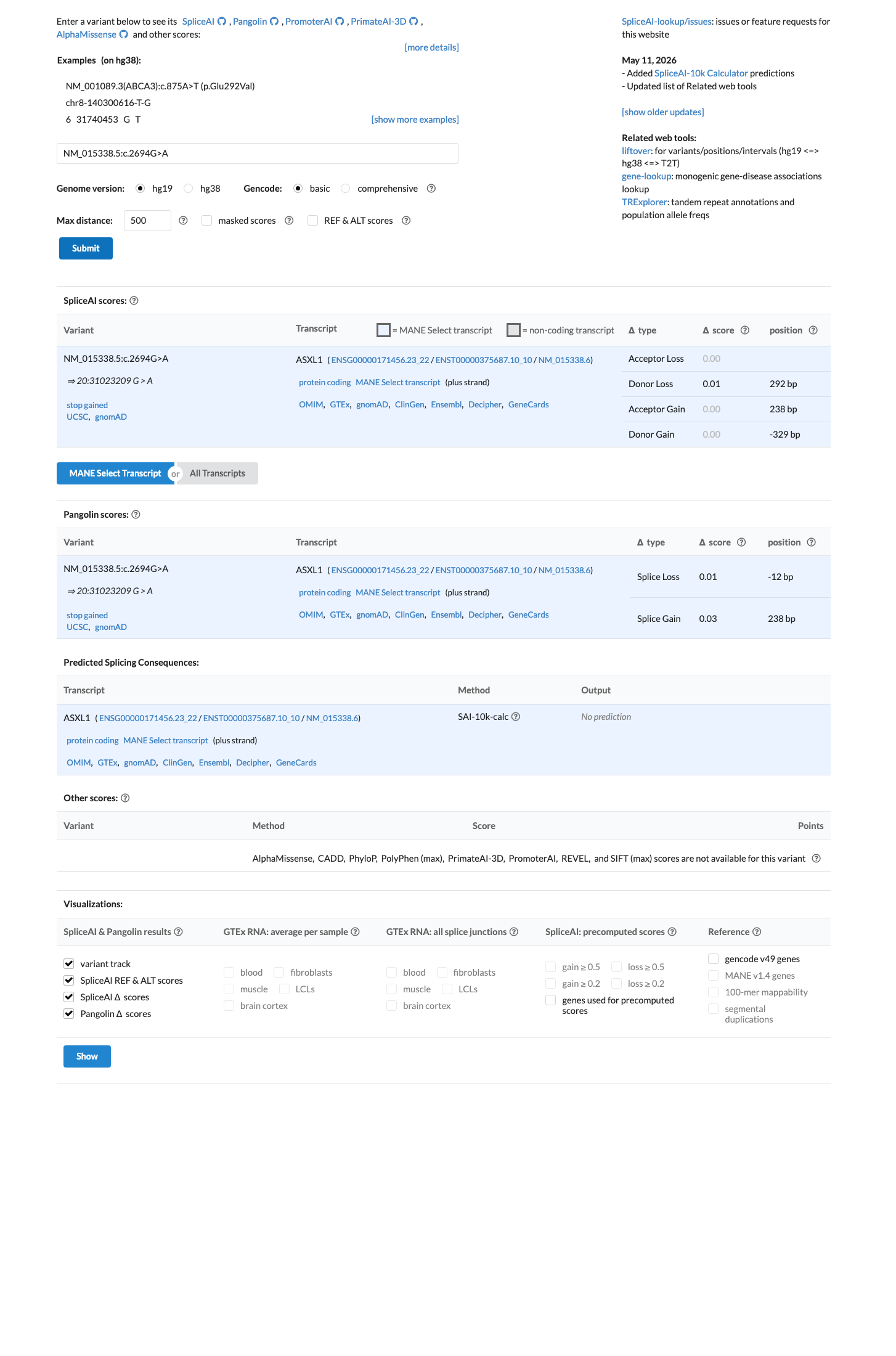
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). BayesDel score = 0.66.
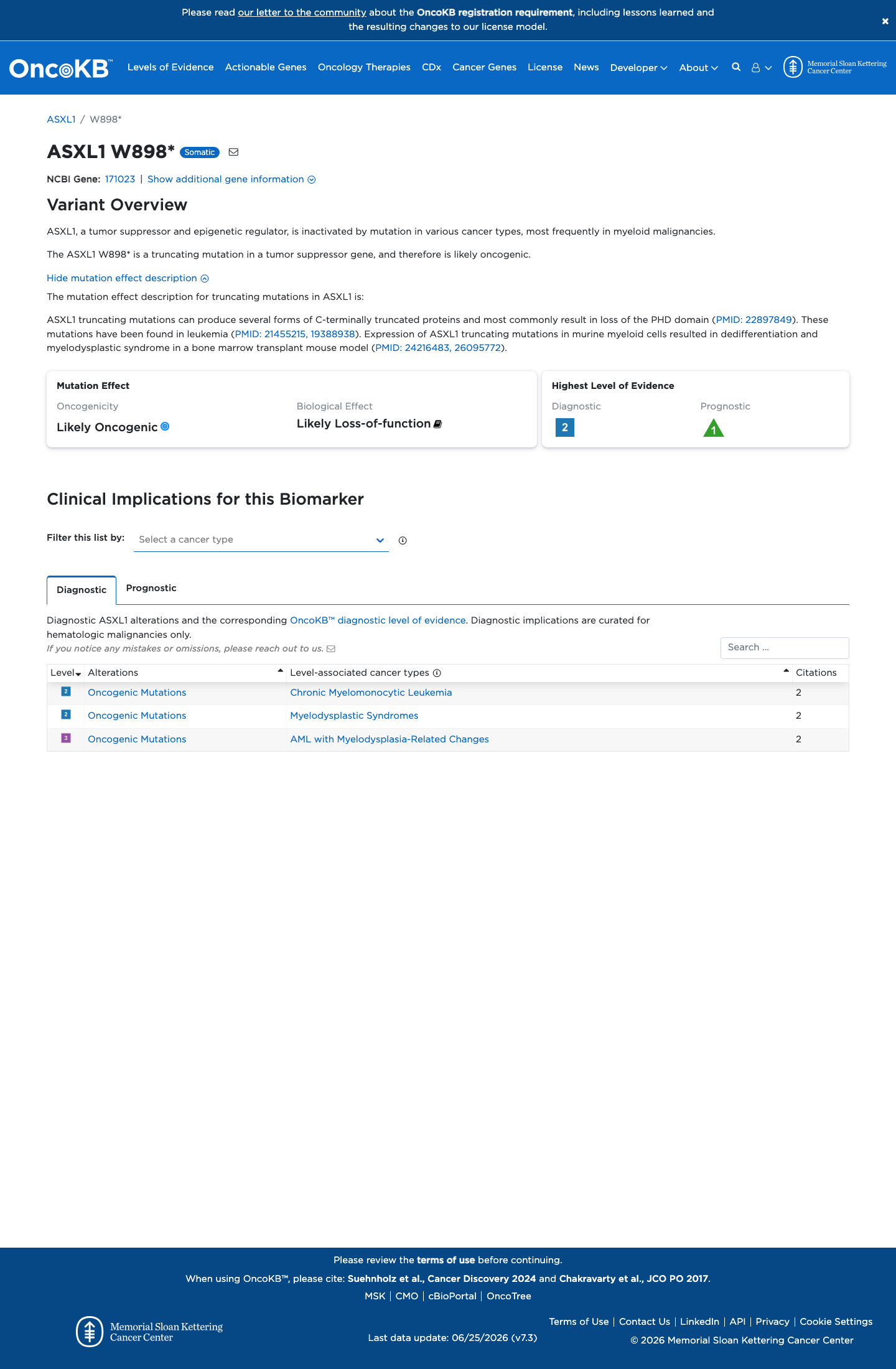
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
19388938 ↗
Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia.
ONCOKB
21455215 ↗
Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms.
ONCOKB
22897849 ↗
ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression.
ONCOKB
24216483 ↗
Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations.
ONCOKB
26095772 ↗
Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex.
ONCOKB