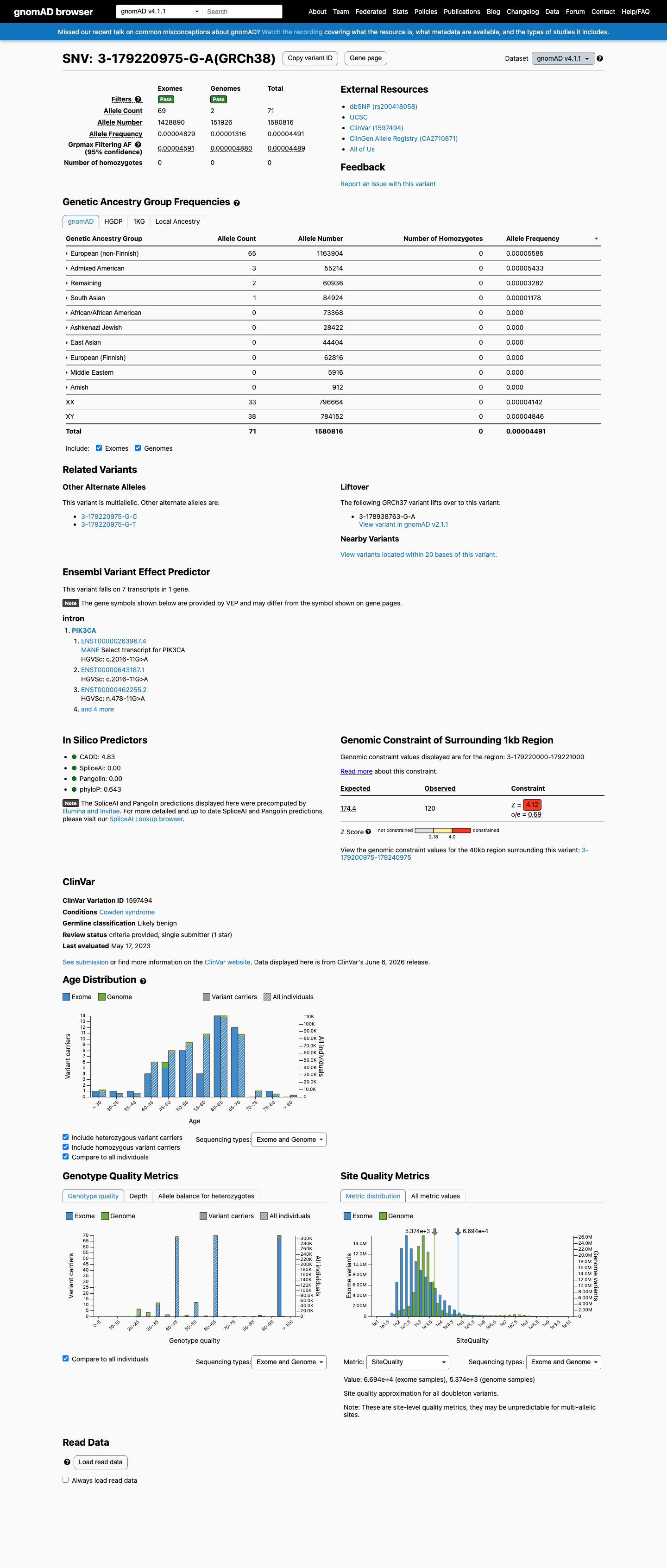
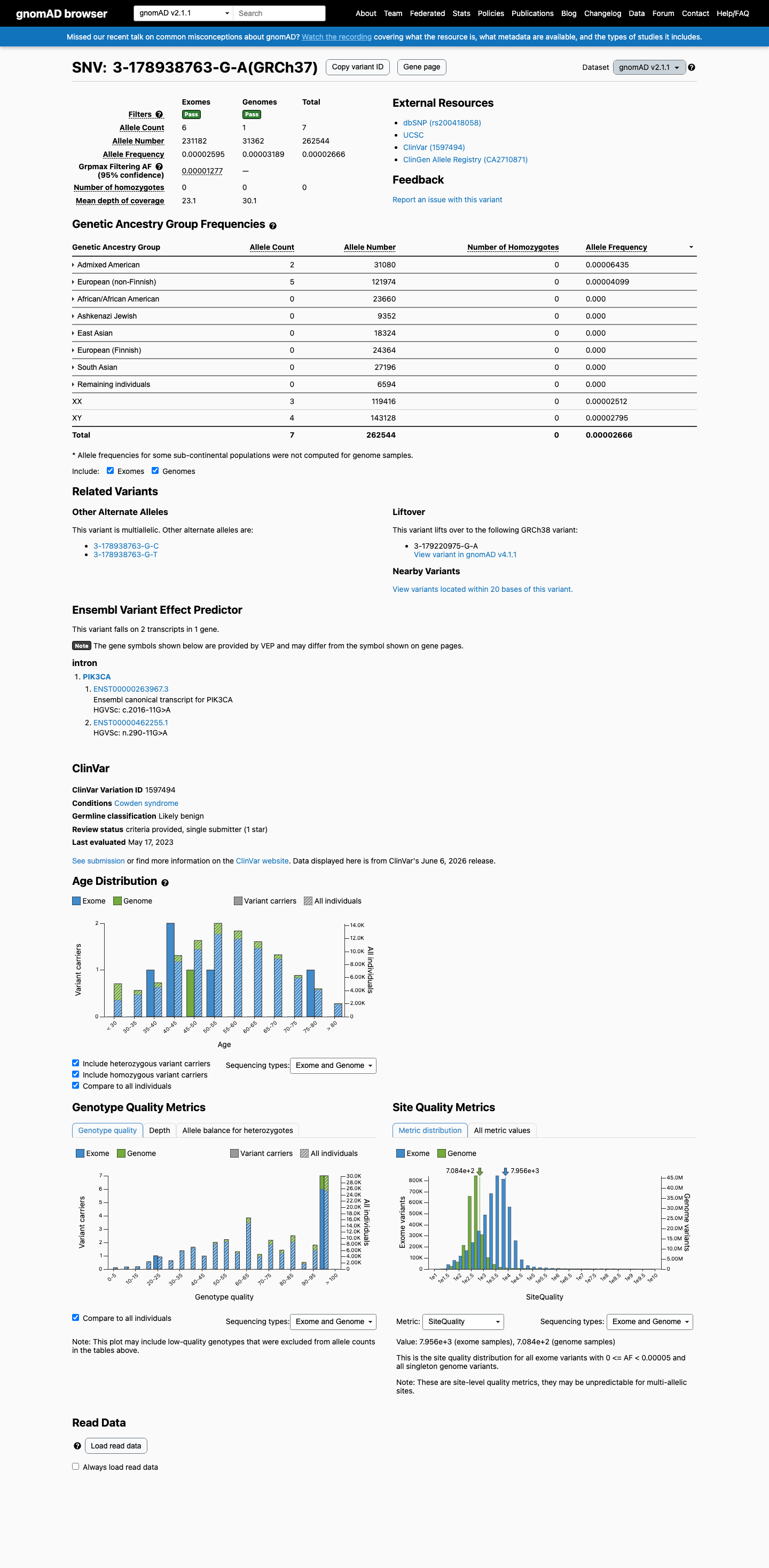
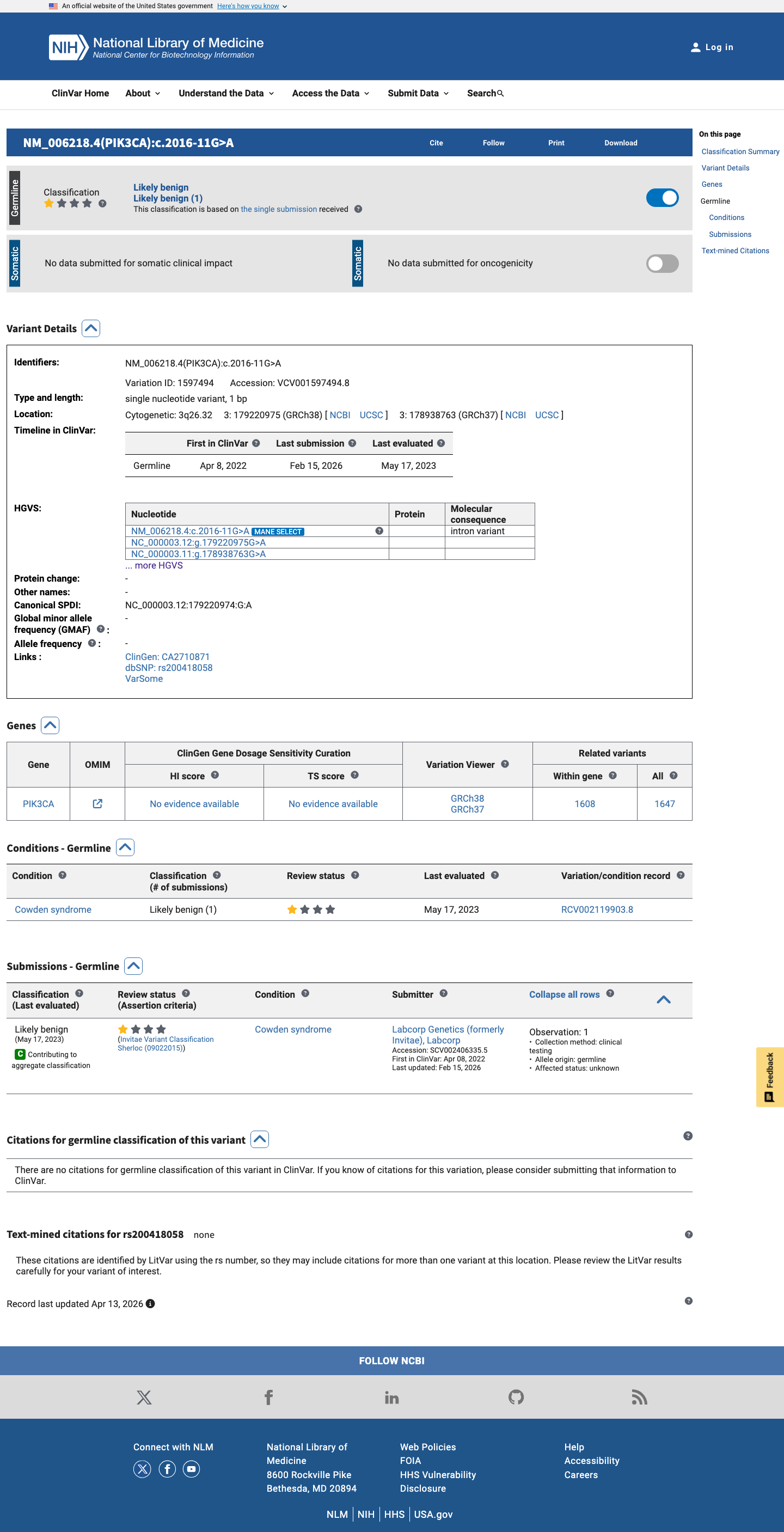
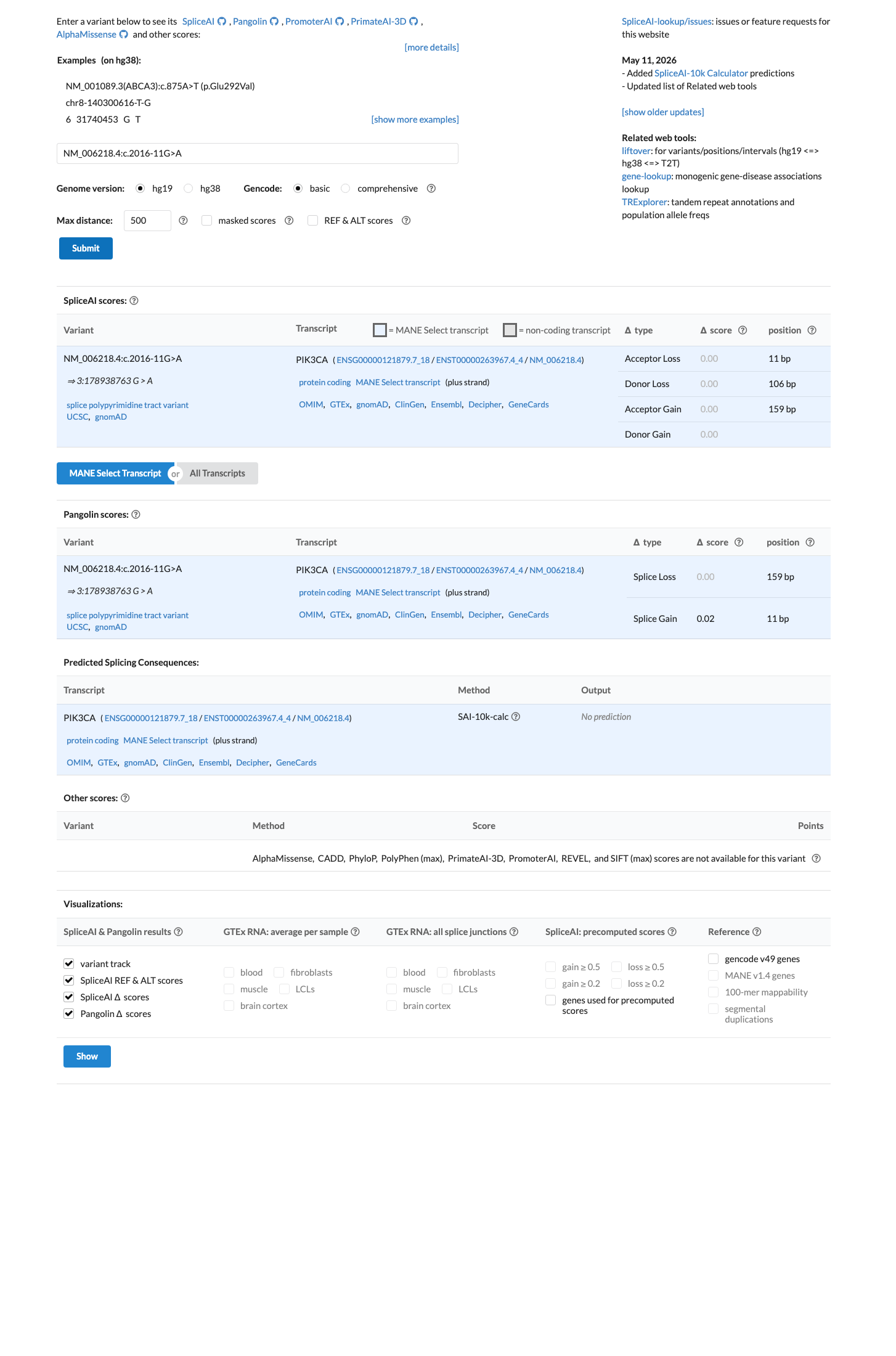
NM_006218.4:c.2016-11G>A is an intronic variant in PIK3CA (intron 13, 11 bases upstream of exon 13). The variant has been observed at low frequency in population databases: 7/262,544 alleles in gnomAD v2.1 (AF 0.00267%) and 71/1,580,816 alleles in gnomAD v4.1 (AF 0.00449%), with no homozygotes.1 SpliceAI predicts no splicing impact (max delta score = 0.00), suggesting the intronic nucleotide substitution does not create or disrupt a splice site.2 This variant has been reported in ClinVar as Likely benign by a single clinical laboratory (Labcorp Genetics, SCV002406335) with review status 'criteria provided, single submitter.' No expert panel classifications are available.3 No functional studies, case reports, segregation data, or de novo observations have been published for this variant. All six associated ClinVar PMIDs are general practice guidelines or background reviews with no variant-specific evidence.4 Under the Brain Malformations VCEP (v1.1.0) Tavtigian point framework, no pathogenic criteria are met and no benign criteria are met. The total point score is 0, which falls within the VUS range (0 to 5 points).5 Multiple benign-suggesting criteria (BP4, BP7) could not be fully assessed due to missing computational data (varSEAK, MaxEntScan, PhyloP). If additional splicing prediction tools confirm no impact and PhyloP indicates low conservation, this variant may be reclassified toward likely benign.6
PIK3CA
Final classification
VUS
PIK3CA c.2016-11G>A · p.?
PIK3CA
NM_006218.4:c.2016-11G>A is an intronic variant in PIK3CA (intron 13, 11 bases upstream of exon 13). The variant has been observed at low frequency in population databases: 7/262,544 alleles in gnomAD v2.1 (AF 0.00267%) and 71/1,580,816 alleles in gnomAD v4.1 (AF 0.00449%), with no homozygotes.
Brain Malformations Specification Tavtigian point framework v1.1.0 point-based framework: no point-contributing criteria = 0 points, which maps to VUS.
Classification rationale
VUS
PIK3CA c.2016-11G>A
Gene diagram
· NM_006218.4 · variants mapped to exon structure
PIK3CA
NM_006218.4
Fetching transcript structure from UCSC…
Applied criteria · 0 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 4.49135e-05; MAF= 0.00449%, 71/1580816 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 5.58465e-05; MAF= 0.00558%, 65/1163904 alleles, homozygotes = 0); grpmax FAF= 4.489e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 2.66622e-05; MAF= 0.00267%, 7/262544 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 6.43501e-05; MAF= 0.00644%, 2/31080 alleles, homozygotes = 0); grpmax FAF= 1.277e-05.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0045%
· 71 / 1,580,816
0 hom · FAF 0.0045%
0 hom · FAF 0.0045%
European (non-Finnish) 65 / 1,163,904 |
0.0056% |
Admixed American 3 / 55,214 |
0.0054% |
Remaining individuals 2 / 60,936 |
0.0033% |
South Asian 1 / 84,924 |
0.0012% |
+ 6 not observed (European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0027%
· 7 / 262,544
0 hom · FAF 0.0013%
0 hom · FAF 0.0013%
Admixed American 2 / 31,080 |
0.0064% |
European (non-Finnish) 5 / 121,974 |
0.0041% |
+ 6 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory). (ClinVarID = 1597494)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
23519317 ↗
Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions.
CLINVAR
26389258 ↗
Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR