NM_003925.3:c.682G>C (p.Val228Leu) is a missense variant in MBD4 exon 3, observed at extremely low frequency in population databases (gnomAD v2.1 AF = 0.011%, 31/282,140 alleles; gnomAD v4.1 AF = 0.0079%, 127/1,614,038 alleles; no homozygotes).1 Multiple in silico tools unanimously predict a benign effect: REVEL score 0.199, BayesDel score −0.381, SpliceAI max delta 0.00, and CADD 7.985. Repo et al. (2020) also reported in silico predictions as polymorphism, benign, tolerated, and no splicing impact.2 This variant has been reported in ClinVar as Likely benign by Ambry Genetics (SCV005618014) and as Uncertain significance by Invitae (SCV003476936). No expert panel review has been performed (ClinVar Variation ID: 2163156).3 The variant was identified in 1 of 440 Finnish patients with uveal melanoma (Repo et al. 2020). The authors considered it likely benign based on in silico predictions and noted no enrichment in cases compared to the Finnish population frequency (0.032%).4 No functional studies, segregation data, de novo observations, or case-control association data are available for this variant. MBD4 is not an established gene where missense variants are a common disease mechanism; the primary pathogenic mechanism is loss of function via truncating variants.5 Based on generic ACMG/AMP 2015 criteria, the evidence profile consists of PM2_supporting (low population frequency) and BP4_supporting (multiple benign in silico predictions). These opposing criteria result in an overall classification of Uncertain significance.6
MBD4
Final classification
VUS
MBD4 c.682G>C · p.Val228Leu
MBD4
NM_003925.3:c.682G>C (p.Val228Leu) is a missense variant in MBD4 exon 3, observed at extremely low frequency in population databases (gnomAD v2.1 AF = 0.011%, 31/282,140 alleles; gnomAD v4.1 AF = 0.0079%, 127/1,614,038 alleles; no homozygotes).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
MBD4 c.682G>C
PM2 + BP4
→
VUS
Gene diagram
· NM_003925.3 · variants mapped to exon structure
MBD4
NM_003925.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 19 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_003925.3:c.682G>C is present at extremely low frequency in population databases: gnomAD v2.1 overall allele frequency is 0.011% (31/282,140 alleles) and gnomAD v4.1 overall allele frequency is 0.0079% (127/1,614,038 alleles), both below the 0.1% PM2 threshold. No homozygotes are observed. The highest subpopulation frequency is in the Finnish population at 0.032% (v2.1), also well below 0.1%.
gnomAD v2.1: AF = 0.011%31/282140 alleles
✓
BP4
supporting
Benign
Multiple lines of computational evidence unanimously predict a benign effect: REVEL score 0.199, BayesDel score −0.381, SpliceAI max delta 0.00, and CADD 7.985. Repo et al. 2020 also reported in silico predictions as polymorphism (MutationTaster), benign (PolyPhen), tolerated (SIFT), and no splicing impact (HSF), supporting BP4 at supporting strength.
REVEL: 0.199 (benign).BayesDel: −0.381 (benign).SpliceAI: max delta 0.00 (no splice impact).
Assessed · not applied
Pathogenic
PS2
No de novo observation of NM_003925.3:c.682G>C has been reported in any reviewed publication or database.
PS3
No variant-specific functional assay for NM_003925.3:c.682G>C (p.Val228Leu) was identified.
PS4
The variant was observed in 1 of 440 Finnish uveal melanoma patients (Repo et al.
PM1
The variant resides in exon 3 of MBD4, within the methyl-CpG binding domain region, but residue Val228 is not located in a statistically significant mutational hotspot, and no functional domain-specific constraint evidence supports PM1.
PM6
No de novo observation of NM_003925.3:c.682G>C has been reported; no maternity/paternity confirmation data exist for this variant.
PP1
No family segregation data are available for NM_003925.3:c.682G>C in any reviewed publication or database.
PP2
MBD4 is not a gene in which missense variants are a recognized common mechanism of disease.
PP3
Multiple in silico tools unanimously predict a benign effect: REVEL score 0.199 (below the 0.29 benign threshold), BayesDel score −0.381 (negative scores favor benign), SpliceAI max delta 0.00 (no splicing impact), and CADD 7.985 (below common pathogenicity thresholds).
PP4
No specific patient phenotype data for NM_003925.3:c.682G>C carriers are available that would allow evaluation of phenotype specificity.
PP5
No reputable source has independently classified NM_003925.3:c.682G>C as pathogenic.
Benign
BA1
The overall allele frequency in gnomAD v2.1 is 0.011%, well below the BA1 threshold of >1%.
BS1
The overall allele frequency in gnomAD v2.1 is 0.011%, well below the BS1 threshold of >0.3%.
BS2
No evidence of observation in healthy adult controls specifically documented for this variant.
BS3
No functional studies demonstrating a benign effect of NM_003925.3:c.682G>C (p.Val228Leu) have been performed.
BS4
No segregation data are available for NM_003925.3:c.682G>C.
BP1
Although MBD4 disease mechanism is primarily loss of function via truncating variants, the missense variant NM_003925.3:c.682G>C cannot automatically be considered benign under BP1.
BP2
No observation of NM_003925.3:c.682G>C in trans with a known pathogenic MBD4 variant has been reported.
BP5
No alternate molecular basis for disease has been identified in individuals carrying NM_003925.3:c.682G>C.
BP6
No reputable source has independently classified NM_003925.3:c.682G>C as benign.
N/A · 7
PVS1 · PS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency

gnomAD v4.1
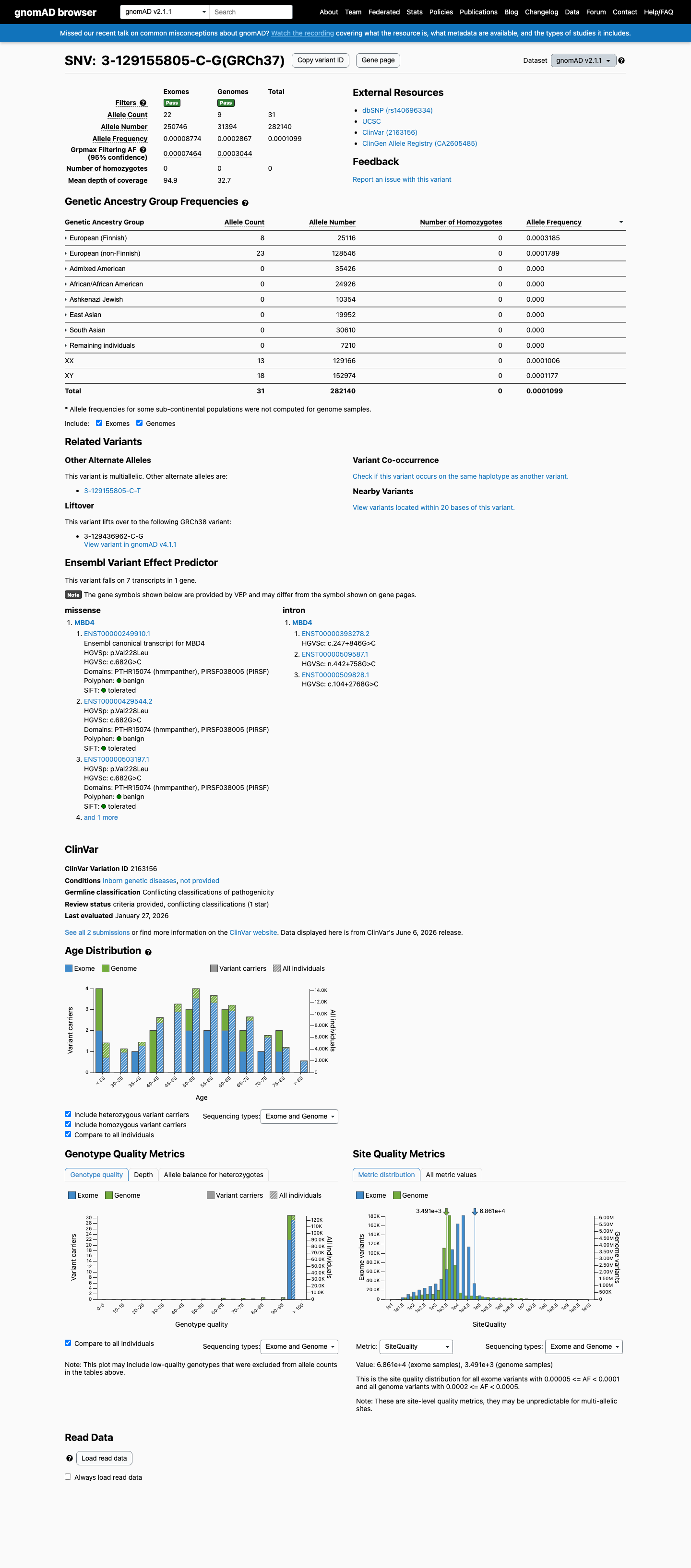
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 7.86846e-05; MAF= 0.00787%, 127/1614038 alleles, homozygotes = 0) and has highest observed frequency in the European (Finnish) population (AF= 0.000296884; MAF= 0.02969%, 19/63998 alleles, homozygotes = 0); grpmax FAF= 7.573e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000109875; MAF= 0.01099%, 31/282140 alleles, homozygotes = 0) and has highest observed frequency in the European (Finnish) population (AF= 0.000318522; MAF= 0.03185%, 8/25116 alleles, homozygotes = 0); grpmax FAF= 0.00030441.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0079%
· 127 / 1,614,038
0 hom · FAF 0.0076%
0 hom · FAF 0.0076%
European (Finnish) 19 / 63,998 |
0.03% |
European (non-Finnish) 106 / 1,180,050 |
0.009% |
Remaining individuals 2 / 62,488 |
0.0032% |
+ 7 not observed (Admixed American, Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.011%
· 31 / 282,140
0 hom · FAF 0.03%
0 hom · FAF 0.03%
European (Finnish) 8 / 25,116 |
0.032% |
European (non-Finnish) 23 / 128,546 |
0.018% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
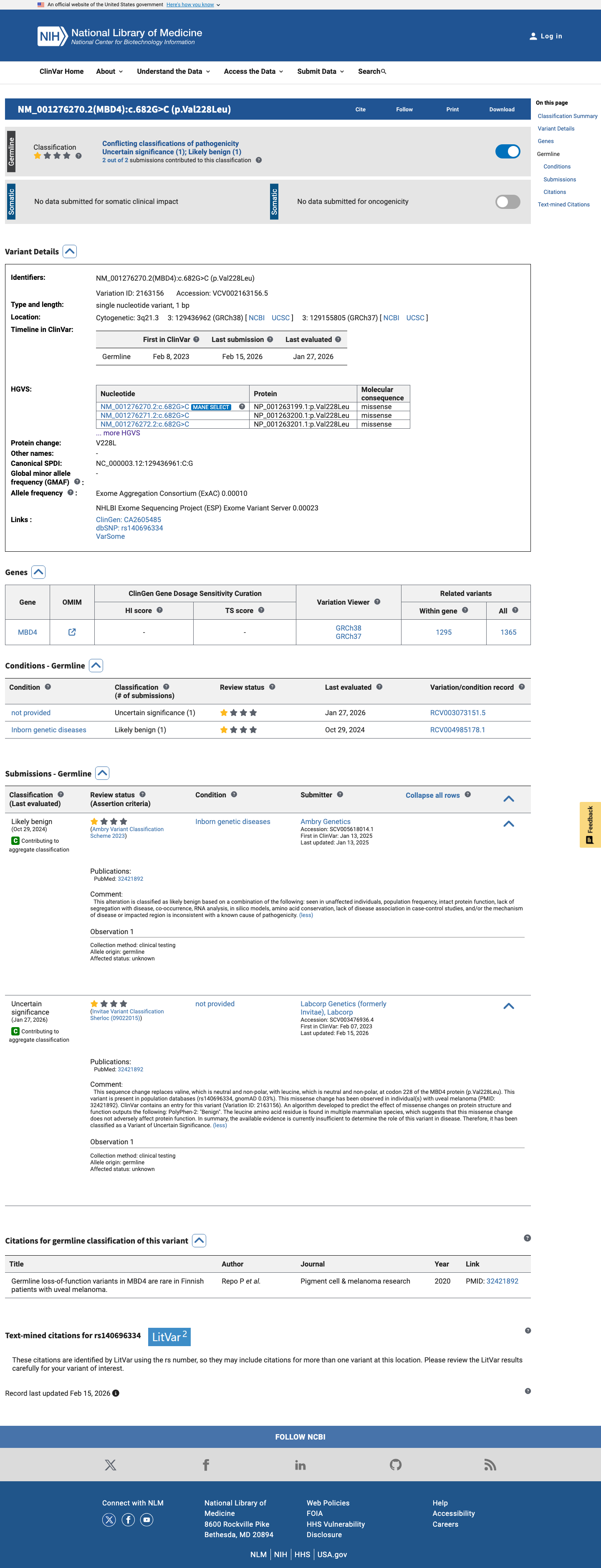
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory) and as Uncertain significance (1 clinical laboratory). (ClinVarID = 2163156)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.199. BayesDel score = -0.380622.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MBD4, a DNA glycosylase, is infrequently altered in cancer.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
25730230 ↗
Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine.
CLINVAR
32421892 ↗
Germline loss-of-function variants in MBD4 are rare in Finnish patients with uveal melanoma.
CLINVAR
23037933 ↗
Including the initial newborn screening bloodspot collection device serial number on birth certificates: basis and recommendations from the Secretary of Health and Human Services' Advisory Committee on Heritable Disorders in Newborns and Children.
CLINVAR
31022120 ↗
ACOG Committee Opinion No. 778 Summary: Newborn Screening and the Role of the Obstetrician-Gynecologist.
CLINVAR