NM_000251.3:c.1316_1318del is an in-frame deletion of 3 nucleotides in exon 8 of MSH2, resulting in p.Pro439del. This variant is absent from all population databases including gnomAD v2.1, v4.1, and gnomAD-Canada, meeting the InSiGHT MSH2 VCEP v2.0.0 PM2_Supporting criterion.1 The InSiGHT MSH2 VCEP v2.0.0 declares multiple criteria as not applicable for MMR gene classification: PM1 (no recognized mutational hotspots), PM4 (protein length change from in-frame variants not used), PS4 (IHC data preferred over proband counting), PP5, BP6, BP1, BP2, BP3, PM6, and PP2.2 PVS1 is not applicable to this in-frame deletion under VCEP rules as it does not introduce a premature termination codon, does not involve canonical splice sites, and no patient mRNA assay data is available. PS1 and PM5 are not applicable as they are defined for missense substitutions only.3 PP3 and BP4 are not applicable to this in-frame deletion under VCEP rules, which specify HCI prior thresholds for missense variants and SpliceAI thresholds for intronic/synonymous variants. SpliceAI predicts no splicing impact (max delta = 0.01), consistent with the variant's location within exon 8 and its in-frame nature.4 No functional assay data (PS3/BS3), segregation data (PP1/BS4), tumor phenotype data (PP4/BP5), de novo reports (PS2), or trans co-occurrence data (BS2) are available for this variant. The ClinVar entry (Variation ID 90623) reports classifications of Uncertain significance by 4 clinical laboratories and Likely pathogenic by 2 laboratories, with review status 'criteria provided, single submitter' at the star-level aggregate.5 Eight publications were reviewed for this variant, including full-text analysis where available. None of the reviewed publications mention NM_000251.3:c.1316_1318del specifically. The publications include methodological papers (SIFT Indel), ACMG/AMP guideline documents, Lynch syndrome management guidelines, and hereditary cancer syndrome reviews.6 Based on the InSiGHT MSH2 VCEP v2.0.0 framework, the only applicable criterion met is PM2_Supporting (absent from gnomAD). With a single supporting pathogenic criterion and no benign criteria, the evidence is insufficient for classification beyond Variant of Uncertain Significance per the VCEP combination rules. Additional evidence from tumor studies, segregation analysis, or functional assays would be required to reach a more definitive classification.7
MSH2
Final classification
VUS
MSH2 c.1316_1318del · p.Pro439del
MSH2
NM_000251.3:c.1316_1318del is an in-frame deletion of 3 nucleotides in exon 8 of MSH2, resulting in p.Pro439del. This variant is absent from all population databases including gnomAD v2.1, v4.1, and gnomAD-Canada, meeting the InSiGHT MSH2 VCEP v2.0.0 PM2_Supporting criterion.
Richards et.al., 2015 - Combining rules v2.0.0 criteria-combination framework was evaluated deterministically with applied criteria: PM2 supporting; no rule matched the adjudicated criteria.
Classification rationale
PM2
VUS
MSH2 c.1316_1318del
PM2
→
VUS
Gene diagram
· NM_000251.3 · variants mapped to exon structure
MSH2
NM_000251.3
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 11 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000251.3:c.1316_1318del is absent from gnomAD v2.1, gnomAD v4.1, and gnomAD-Canada v1.0. This meets the InSiGHT MSH2 VCEP v2.0.0 PM2_Supporting threshold of absent/extremely rare allele frequency <0.00002 (<1 in 50,000 alleles) in gnomAD v4.
Absent from gnomAD v2.1 (0 alleles)Absent from gnomAD v4.1 (0 alleles)Absent from gnomAD-Canada v1.0 (0 alleles)
Assessed · not applied
Pathogenic
PVS1
NM_000251.3:c.1316_1318del is an in-frame deletion (p.Pro439del) in exon 8 of MSH2.
PS2
No de novo occurrence data is available for NM_000251.3:c.1316_1318del.
PS3
No variant-specific functional assay data is available for NM_000251.3:c.1316_1318del.
PP1
No co-segregation data is available for NM_000251.3:c.1316_1318del.
PP4
No tumor MSI/IHC data is available for patients carrying NM_000251.3:c.1316_1318del.
Benign
BA1
BA1 requires gnomAD v4 Grpmax filtering allele frequency >=0.001 (0.1%).
BS1
BS1 requires gnomAD v4 Grpmax filtering allele frequency >=0.0001 and <0.001 (0.01-0.1%).
BS2
No data is available on co-occurrence of NM_000251.3:c.1316_1318del in trans with a known pathogenic MSH2 variant.
BS3
No variant-specific functional assay data demonstrating normal protein function is available for NM_000251.3:c.1316_1318del.
BS4
No segregation data is available to evaluate lack of co-segregation with disease.
BP5
No data is available on the presence of an alternate molecular basis for disease in patients carrying NM_000251.3:c.1316_1318del.
N/A · 16
PS1 · PS4 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP3 · PP5 · BP1 · BP2 · BP3 · BP4 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
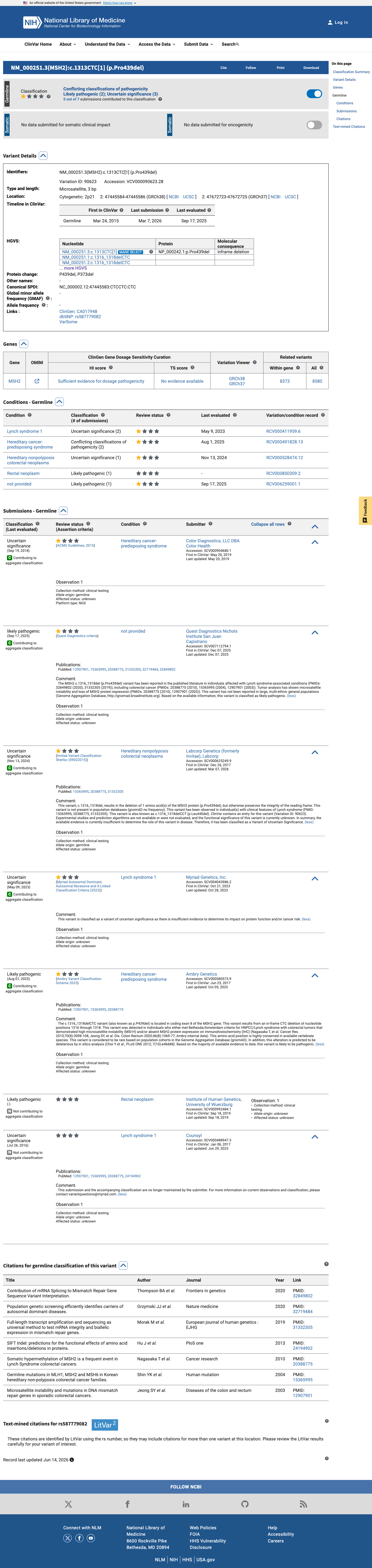
This variant has been reported in ClinVar as Uncertain significance (4 clinical laboratories) and as Likely pathogenic (2 clinical laboratories) and as likely pathogenic (1 clinical laboratory). (ClinVarID = 90623)
In silico
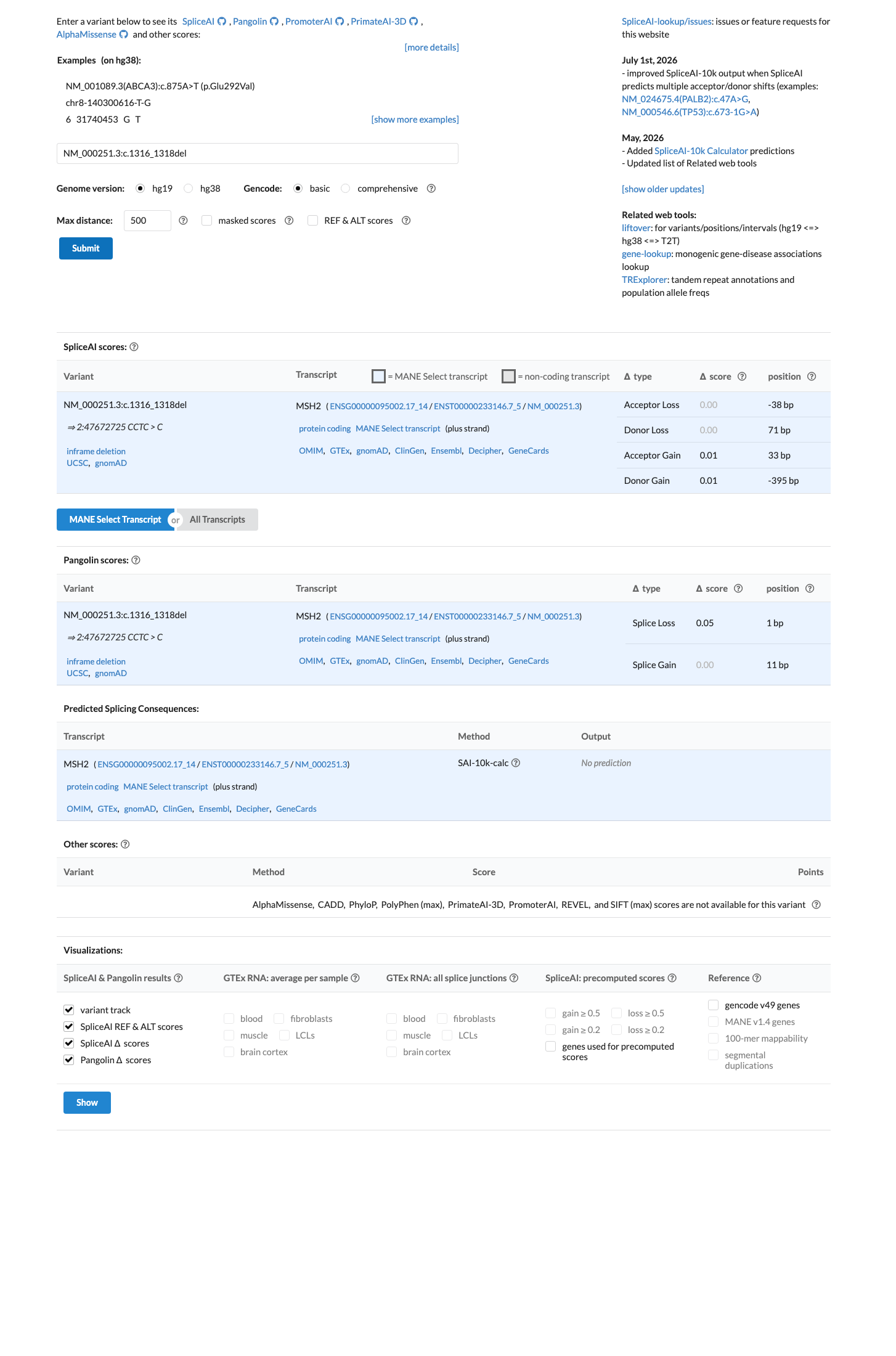
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01).
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MSH2, a DNA mismatch repair protein, is frequently mutated in colorectal, small bowel, and endometrial cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 8 PMIDs not cited in assessment
12907901 ↗
Microsatellite instability and mutations in DNA mismatch repair genes in sporadic colorectal cancers.
CLINVAR
24194902 ↗
SIFT Indel: predictions for the functional effects of amino acid insertions/deletions in proteins.
CLINVAR
25645574 ↗
ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
11598466 ↗
Practice parameters for the identification and testing of patients at risk for dominantly inherited colorectal cancer--supporting documentation.
CLINVAR
23408351 ↗
Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts.
CLINVAR
23535968 ↗
Informing family members of individuals with Lynch syndrome: a guideline for clinical geneticists.
CLINVAR