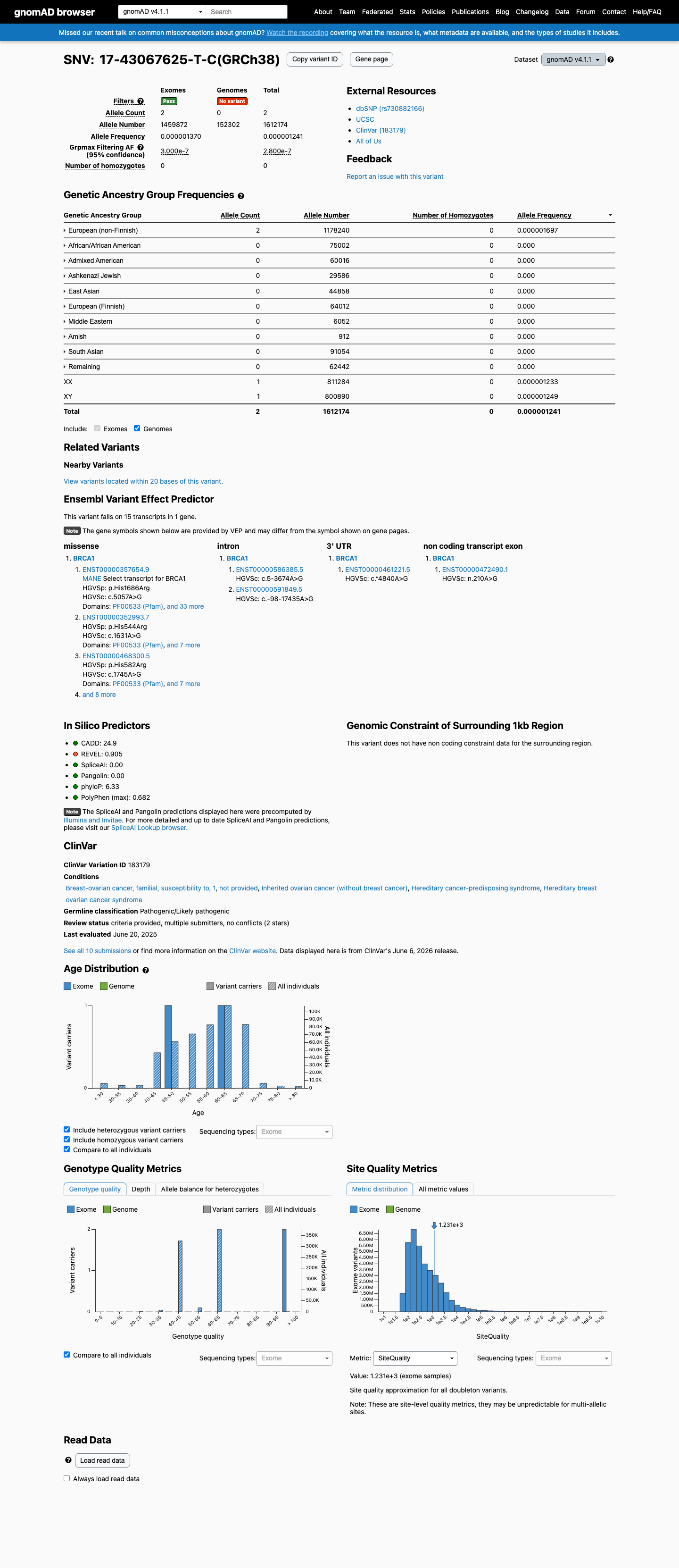
NM_007294.4:c.5057A>G (p.His1686Arg) is a missense variant in BRCA1 exon 16, located within the BRCT repeat domain (aa 1650-1857), a clinically important functional domain critical for BRCA1 tumor suppressor activity.1 The variant meets PS3_Strong per ENIGMA Specifications Table 9, based on three calibrated functional studies (Findlay 2018 PMID:30209399; Petitalot 2019 PMID:30257991; Bouwman 2020 PMID:32546644) that demonstrate protein function similar to pathogenic control variants, with complete functional impact (Loss-of-Function) confirmed in the ST4 functional assay dataset.2 The variant meets PP3_Supporting: it is a missense substitution within the BRCT clinically important functional domain with a BayesDel no-AF score of 0.378 (>=0.28 threshold), predicting deleterious impact via protein change. REVEL score is 0.905. SpliceAI max delta is 0.15, indicating no significant splicing impact.3 The variant meets PM2_Supporting: it is absent from gnomAD v2.1 (non-cancer, exome only) and present at ultra-rare frequency in gnomAD v4.1 (2/1,612,174 alleles; grpmax FAF=2.8e-07). It is absent from gnomAD-Canada v1.0.4 PP4 could not be applied: the clinical-history likelihood ratio is LR=1.42 (1 proband, Li et al. 2020 PMID:31853058), which falls in the ENIGMA neutral zone (0.48-2.08) and provides insufficient evidence in either direction.5 The variant does not meet PVS1 (missense, not a null variant), PS1 (no different nucleotide to same amino acid comparator), PS4 (no case-control study), PP1 (no co-segregation data), BA1/BS1 (frequency too low), BS2 (no healthy adult data), BS3 (functional evidence shows damaging effect), BS4 (no lack-of-segregation data), BP1 (inside functional domain), BP4 (BayesDel >0.15, SpliceAI >0.1), BP5 (LR neutral zone), or BP7 (damaging functional effect).6
BRCA1
Final classification
Likely Pathogenic
BRCA1 c.5057A>G · p.His1686Arg
BRCA1
NM_007294.4:c.5057A>G (p.His1686Arg) is a missense variant in BRCA1 exon 16, located within the BRCT repeat domain (aa 1650-1857), a clinically important functional domain critical for BRCA1 tumor suppressor activity.
ENIGMA BRCA1/BRCA2 VCEP v1.2.0 Table 3 combination rules applied to adjudicated criteria: 1 Strong (PS3) + 2 Supporting (PM2, PP3) satisfies the Likely Pathogenic combination rule (1 Strong + ≥2 Supporting). No benign criteria are met, so no conflicting-evidence point-system reduction is required.
Classification rationale
PS3PM2PP3
Likely Pathogenic
BRCA1 c.5057A>G
PS3 + PM2 + PP3
→
Likely Pathogenic
1
cspec ↗
2
vcep_specifications_table9_v1_2_2024_11_18PMID:30209399 ↗vcep_supplementarytables_v1_2_2024_11_18
3
bayesdelrevelspliceai ↗vcep_supplementarytables_v1_2_2024_11_18
5
PMID:31853058 ↗vcep_pmid_31853058_brca1_clinical_history_lr
6
cspec ↗gnomad_v4 ↗bayesdelspliceai ↗
Gene diagram
· NM_007294.4 · variants mapped to exon structure
BRCA1
NM_007294.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 14 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Per ENIGMA Specifications Table 9, NM_007294.4:c.5057A>G (p.His1686Arg) is assigned PS3_Strong based on three calibrated functional studies that demonstrate protein function similar to pathogenic control variants: Findlay 2018 (PMID:30209399) saturation genome editing — classified as Loss-of-Function; Petitalot 2019 (PMID:30257991) — classified as 3P (pathogenic); and Bouwman 2020 (PMID:32546644) — classified as Deleterious. The variant shows complete functional impact (LOF, score -0.447) in the ST4 functional assay dataset and is annotated as LossOfFunction in ST13 with three supporting calibrated studies. SpliceAI max delta = 0.15 indicates no predicted splicing effect, confirming the functional defect is mediated at the protein level.
ENIGMA Table 9 assigns PS3_Strong with three calibrated studiesFindlay 2018 (PMID:30209399): saturation genome editing — Loss-of-FunctionPetitalot 2019 (PMID:30257991): functional assay — 3P (pathogenic classification)
✓
PM2
supporting
Pathogenic
NM_007294.4:c.5057A>G is absent from gnomAD v2.1 (non-cancer, exome only subset), meeting the ENIGMA PM2_Supporting criterion for absence from outbred population controls. In gnomAD v4.1, the variant is present at ultra-rare frequency (2/1,612,174 alleles; grpmax FAF=2.8e-07; highest in European non-Finnish at 2/1,178,240). The variant is also absent from gnomAD-Canada v1.0.
Absent from gnomAD v2.1 (non-cancerexome only subset)gnomAD v4.1: 2/1
✓
PP3
supporting
Pathogenic
NM_007294.4:c.5057A>G (p.His1686Arg) is a missense variant located within the BRCT repeat domain (aa 1650-1857), a clinically important functional domain per ENIGMA specifications. The BayesDel no-AF score is 0.378, which exceeds the ENIGMA PP3 threshold of >=0.28 for predicted impact via protein change. REVEL score is 0.905, consistent with a deleterious prediction. SpliceAI max delta score is 0.15 (<0.2), indicating no significant predicted splicing impact.
Missense variant inside BRCT domain (aa 1686within 1650-1857 clinically important functional domain)BayesDel no-AF score = 0.378 >= 0.28 threshold
Assessed · not applied
Pathogenic
PVS1
NM_007294.4:c.5057A>G is a missense variant (p.His1686Arg) in BRCA1 exon 16.
PS1
No different nucleotide change at codon 1686 producing the same amino acid substitution (His1686Arg) has been classified as pathogenic.
PS4
No case-control study demonstrating significantly increased prevalence of NM_007294.4:c.5057A>G in affected individuals versus controls (p-value <= 0.05 and OR >= 4, lower CI excluding 2.0) was identified.
PP1
No co-segregation data are available for NM_007294.4:c.5057A>G.
PP4
The clinical-history likelihood ratio for NM_007294.4:c.5057A>G is LR = 1.42 (based on 1 proband, from Li et al.
Benign
BA1
The grpmax filter allele frequency (FAF) for NM_007294.4:c.5057A>G in gnomAD v4.1 is 2.8e-07 (0.000028%), which is far below the ENIGMA BA1 threshold of FAF > 0.1% (0.001).
BS1
The grpmax FAF of 2.8e-07 is below both the ENIGMA BS1_Strong threshold (FAF > 0.0001, i.e., >0.01%) and the BS1_Supporting threshold (FAF > 0.00002 and <= 0.0001, i.e., >0.002% and <=0.01%).
BS2
No data are available for observation of NM_007294.4:c.5057A>G in healthy adults without features of Fanconi Anemia.
BS3
ENIGMA Specifications Table 9 assigns PS3_Strong, not BS3, for NM_007294.4:c.5057A>G.
BS4
No quantitative co-segregation analysis demonstrating lack of segregation with disease (LR <= 0.48 for BS4_Supporting) has been performed for NM_007294.4:c.5057A>G.
BP1
NM_007294.4:c.5057A>G (p.His1686Arg) is a missense variant located within the BRCT repeat domain (aa 1650-1857), which is a clinically important functional domain per ENIGMA specifications.
BP4
BP4_Supporting requires: (a) missense variant inside a clinically important functional domain, AND (b) BayesDel no-AF score <= 0.15, AND (c) SpliceAI <= 0.1.
BP5
The clinical-history likelihood ratio for NM_007294.4:c.5057A>G is LR = 1.42 (based on 1 proband).
BP7
NM_007294.4:c.5057A>G is a missense variant.
N/A · 8
PS2 · PM1 · PM5 · PM6 · PP2 · PP5 · BP2 · BP6
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.24056e-06; MAF= 0.00012%, 2/1612174 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.69745e-06; MAF= 0.00017%, 2/1178240 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00012%
· 2 / 1,612,174
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
European (non-Finnish) 2 / 1,178,240 |
0.00017% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
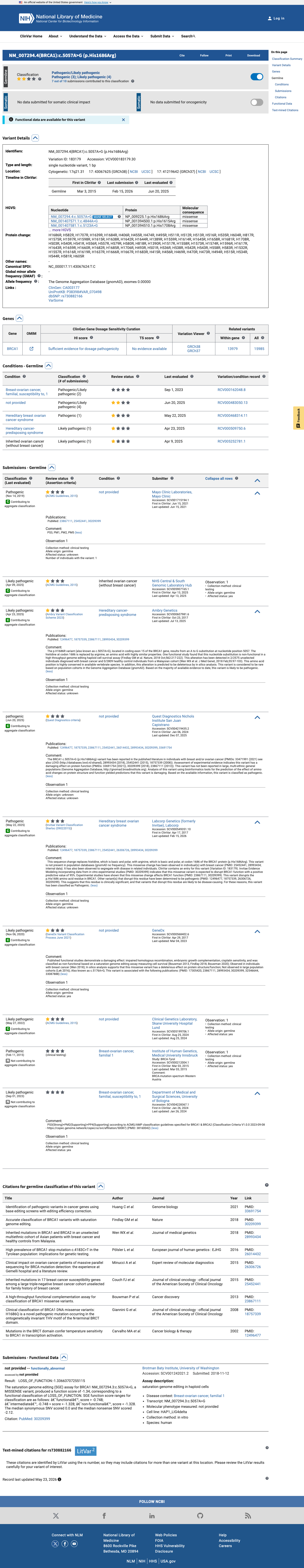
This variant has been reported in ClinVar as Likely pathogenic (5 clinical laboratories) and as Pathogenic (3 clinical laboratories) and as pathogenic (1 clinical laboratory). (ClinVarID = 183179)
In silico
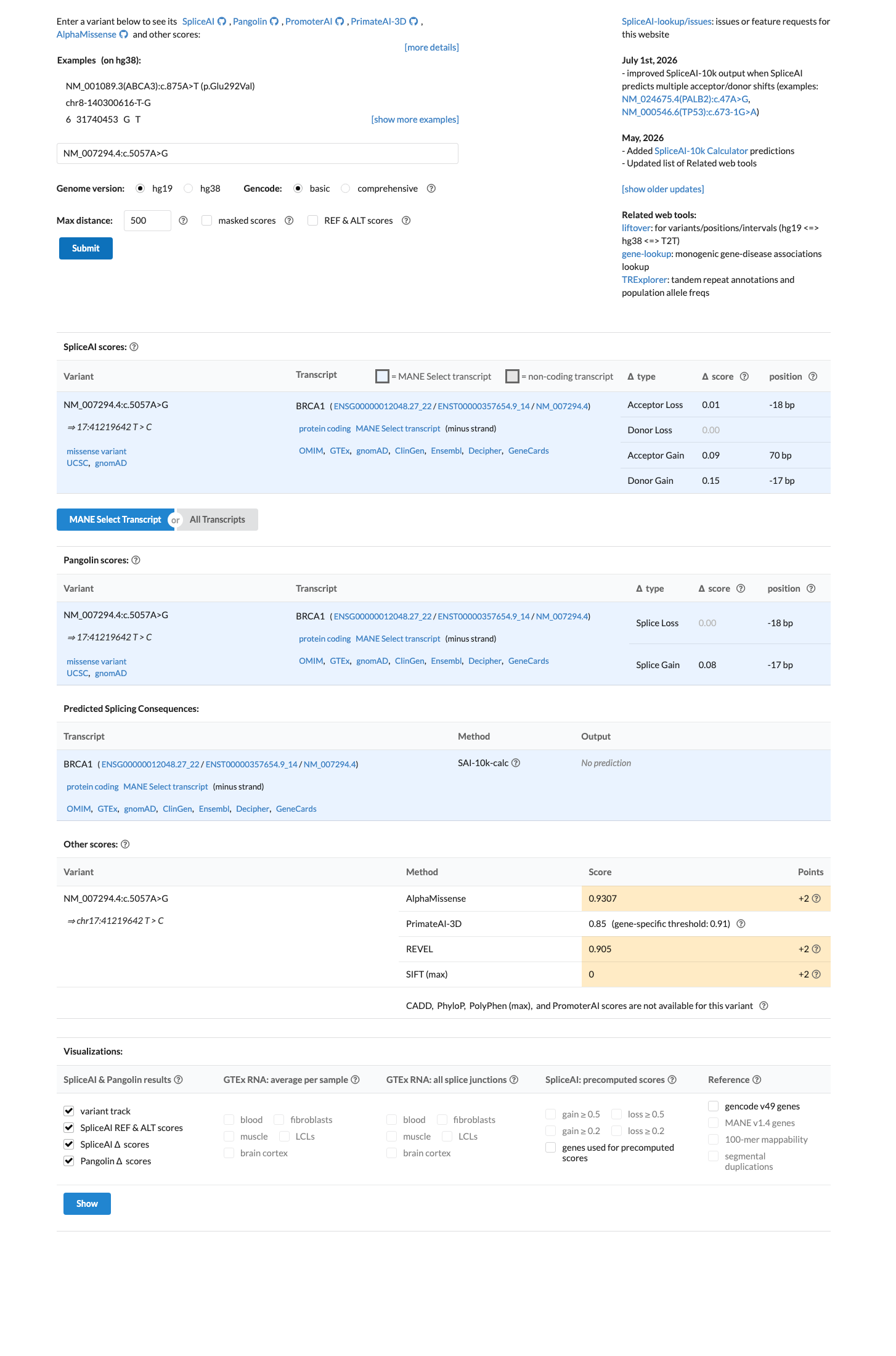
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.15). REVEL score = 0.905. BayesDel score = 0.378152.
Functional
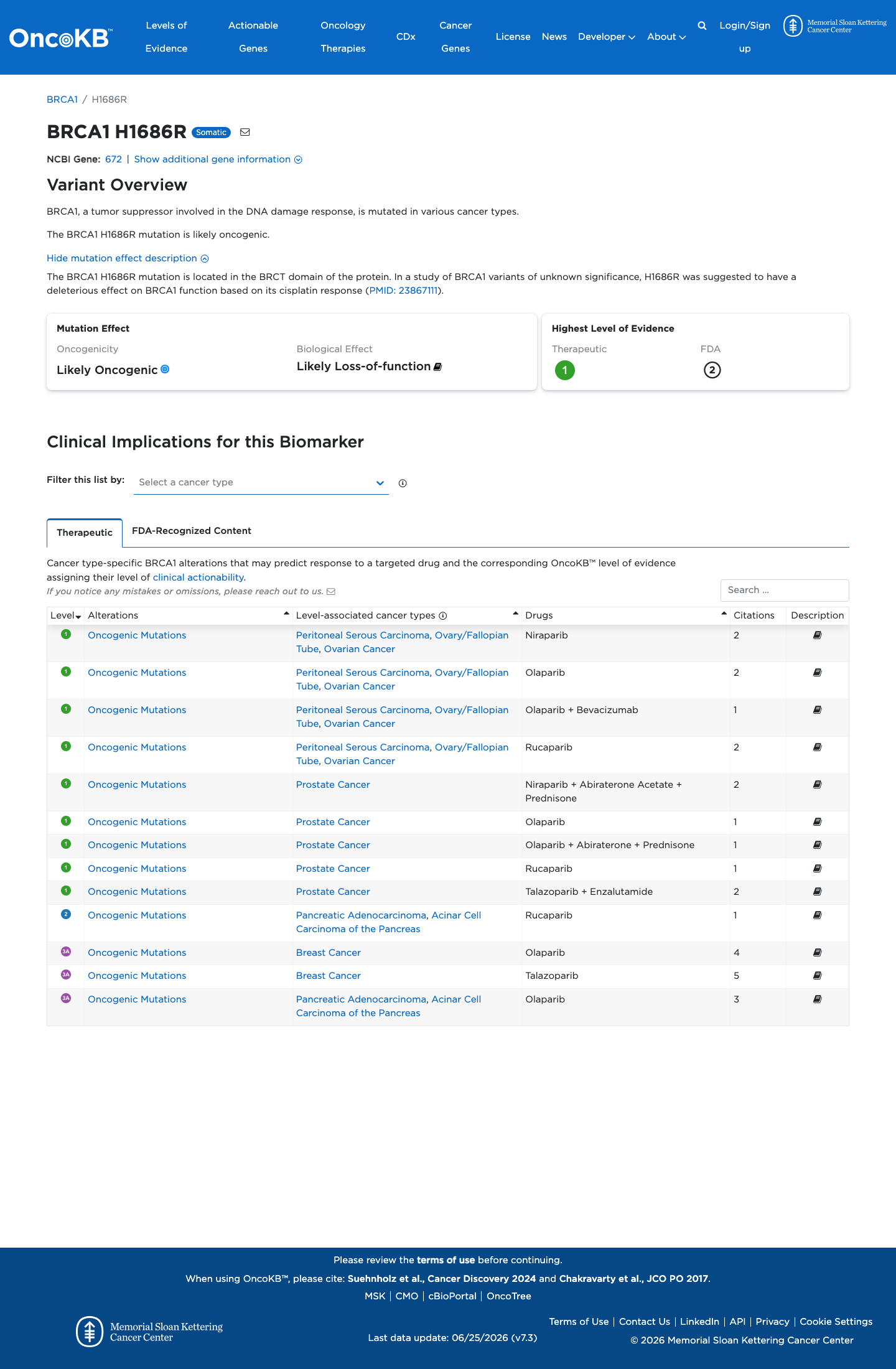
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Accurate classification of BRCA1 variants with saturation genome editing.
Searched
c.5057A>G5057H1686RHis1686Arg1686
Found
Findlay et al. 2018 performed saturation genome editing of BRCA1 RING and BRCT domain exons, functionally classifying 3,893 SNVs. The variant NM_007294.4:c.5057A>G (p.His1686Arg) was among the variants assayed but is listed only in supplementary tables, not in the main text. The ENIGMA VCEP curated the supplementary data and determined the variant exhibits complete Loss-of-Function.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PS3 supports · met
Why
Variant-specific functional score classified as Loss-of-Function in ENIGMA Table 9 and ST4/ST13 datasets; supports PS3_Strong assignment.
Location Supplementary Table 1 (online at sge.gs.washington.edu/BRCA1); main text describes methodology for all BRCT domain variants in exon 16 · Context Saturation genome editing in HAP1 haploid cells, survival-based assay measuring HDR pathway function · full text
Sources & reference links
9Sources
Triaged references · 6 PMIDs not cited in assessment
23867111 ↗
A high-throughput functional complementation assay for classification of BRCA1 missense variants.
ONCOKB
12496477 ↗
Mutations in the BRCT domain confer temperature sensitivity to BRCA1 in transcription activation.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26306726 ↗
Clinical impact on ovarian cancer patients of massive parallel sequencing for BRCA mutation detection: the experience at Gemelli hospital and a literature review.
CLINVAR
28993434 ↗
Inherited mutations in BRCA1 and BRCA2 in an unselected multiethnic cohort of Asian patients with breast cancer and healthy controls from Malaysia.
CLINVAR
33691754 ↗
Identification of pathogenic variants in cancer genes using base editing screens with editing efficiency correction.
CLINVAR