NM_000455.5:c.1226G>A (p.Arg409Gln) is a missense variant in STK11, a gene associated with autosomal dominant Peutz-Jeghers syndrome.1 This variant is present at extremely low frequency in population databases: gnomAD v2.1 allele frequency 0.0036% (7/193,536 alleles) and gnomAD v4.1 allele frequency 0.0033% (52/1,587,284 alleles), with no homozygotes observed. PM2 (supporting) is met.2 Multiple computational prediction tools suggest a benign effect: REVEL score 0.122, BayesDel score -0.514, and SpliceAI max delta 0.00. BP4 (supporting) is met.3 A same-residue variant, p.Arg409Trp (R409W), was evaluated in a functional study (PMID:34849607) and was found to retain WT-like kinase activity and p53-mediated transcriptional activation, suggesting that amino acid substitution at this position may not disrupt STK11 function.4 The variant has been reported in ClinVar predominantly as a variant of uncertain significance (11 clinical laboratories), with 2 laboratories classifying as likely benign and 1 as benign. No expert panel review is available.5 This variant is observed in COSMIC in somatic cancers at low frequency (n=1), which does not provide evidence for germline pathogenicity. No variant-specific publications were identified in the reviewed literature. PMID:34849607 evaluated R409W at the same residue but did not directly study R409Q.6 Applying generic ACMG/AMP 2015 classification rules: one supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4) are met. These offset each other, resulting in an overall classification of Uncertain Significance.7
STK11
Final classification
VUS
STK11 c.1226G>A · p.Arg409Gln
STK11
NM_000455.5:c.1226G>A (p.Arg409Gln) is a missense variant in STK11, a gene associated with autosomal dominant Peutz-Jeghers syndrome.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
STK11 c.1226G>A
PM2 + BP4
→
VUS
Gene diagram
· NM_000455.5 · variants mapped to exon structure
STK11
NM_000455.5
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 22 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000455.5:c.1226G>A is present in gnomAD v2.1 at an allele frequency of 0.0036% (7/193,536 alleles, 0 homozygotes) and in gnomAD v4.1 at 0.0033% (52/1,587,284 alleles, 0 homozygotes), well below the 0.1% threshold for PM2 at supporting level. Absent from gnomAD-Canada.
gnomAD v2.1 AF = 0.0036% (7/193536 alleles)<0.1% threshold
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no impact on the gene product. REVEL score is 0.122 (well below the 0.5 pathogenic cutoff), BayesDel score is -0.514 (strongly in the benign range), and SpliceAI predicts no aberrant splicing (max delta = 0.00). All three independent in silico predictors concur that this variant is likely benign.
REVEL = 0.122 (below 0.5 pathogenic thresholdfavors benign)BayesDel noAF = -0.514 (benign range
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant has been reported at the same nucleotide position (c.1226G>A) with the same amino acid change.
PS2
No de novo occurrence of NM_000455.5:c.1226G>A was identified in any reviewed publication or ClinVar submission.
PS3
No well-established functional study directly assessing NM_000455.5:c.1226G>A (p.Arg409Gln) was identified.
PS4
No case-control study demonstrates statistically significant enrichment of NM_000455.5:c.1226G>A in affected individuals versus controls.
PM1
The variant does not lie in a statistically significant mutational hotspot in STK11.
PM5
PM5 candidate harvesting did not identify a confirmed pathogenic missense variant at the same residue (R409) suitable as a comparator.
PM6
No confirmed de novo occurrence of NM_000455.5:c.1226G>A has been reported.
PP1
No segregation data are available for NM_000455.5:c.1226G>A.
PP2
PP2 applies when a missense variant occurs in a gene with a low rate of benign missense variation and where missense variants are a common mechanism of disease.
PP3
Multiple lines of computational evidence suggest a benign effect rather than a deleterious effect.
PP4
No detailed phenotypic data specific to individuals carrying NM_000455.5:c.1226G>A are available.
PP5
No reputable source (expert panel, professional practice guideline, or clinical diagnostic laboratory with comprehensive evidence) classifies NM_000455.5:c.1226G>A as pathogenic.
Benign
BA1
The variant allele frequency in gnomAD (0.0036% v2.1, 0.0033% v4.1) is far below the BA1 threshold of >1% in any population.
BS1
The variant allele frequency in gnomAD (0.0036% v2.1, 0.0033% v4.1) is well below the BS1 threshold of >0.3% in any population.
BS2
No homozygous observations of NM_000455.5:c.1226G>A have been reported in any population database.
BS3
No well-established functional study directly demonstrates that NM_000455.5:c.1226G>A (p.Arg409Gln) has no deleterious effect.
BS4
No segregation data are available to demonstrate lack of co-segregation with disease.
BP1
BP1 applies when a missense variant occurs in a gene where only truncating variants cause disease.
BP2
No observation of NM_000455.5:c.1226G>A occurring in trans with a known pathogenic STK11 variant has been reported.
BP5
BP5 requires that the variant be found in a case with an alternate molecular basis for disease.
BP6
While two clinical laboratories classify NM_000455.5:c.1226G>A as likely benign and one as benign in ClinVar, these are minority opinions (3 of 16 submissions) and no expert panel or reputable source with comprehensive evidence supports a benign classification.
BP7
BP7 applies to synonymous variants with no predicted splice impact.
N/A · 1
PVS1
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
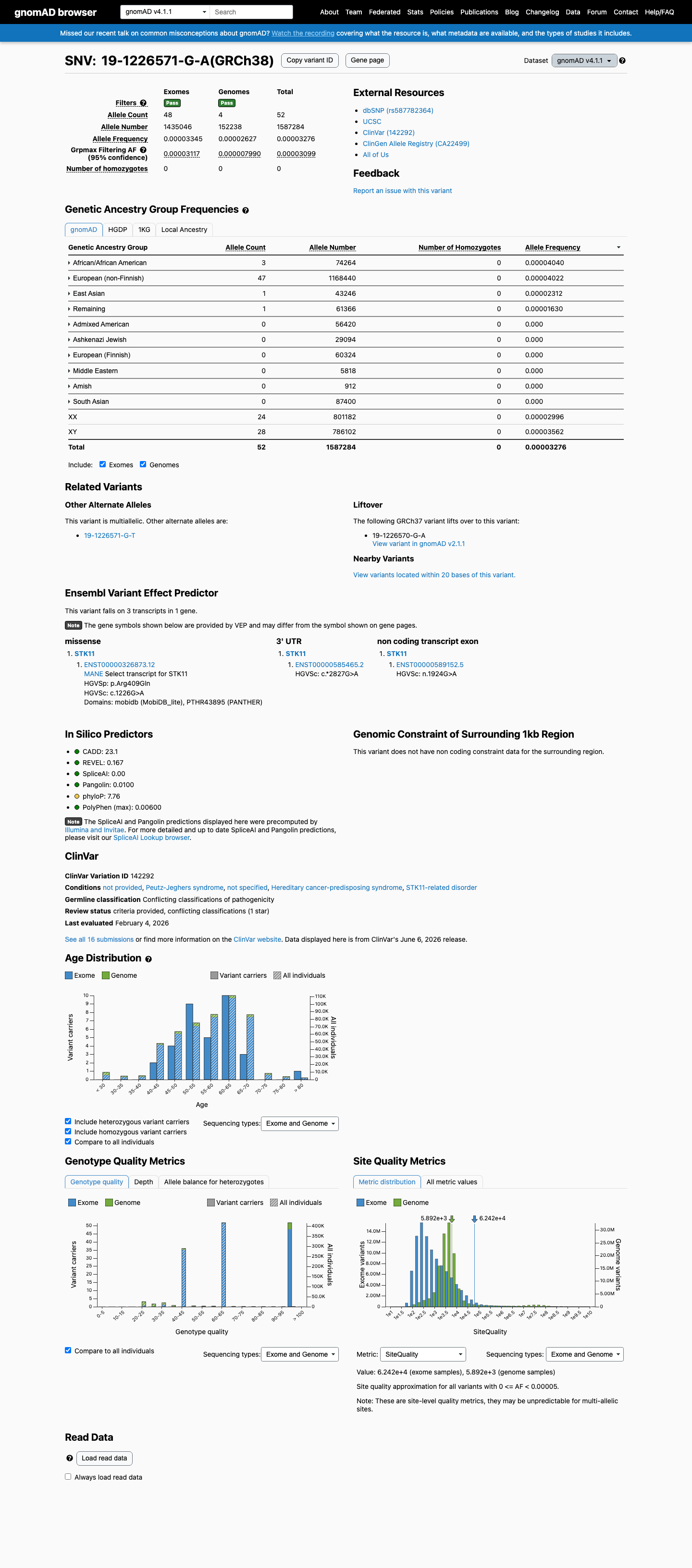
This variant is present in gnomAD v4.1 (AF= 3.27604e-05; MAF= 0.00328%, 52/1587284 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 4.03964e-05; MAF= 0.00404%, 3/74264 alleles, homozygotes = 0); grpmax FAF= 3.099e-05.
v2.1
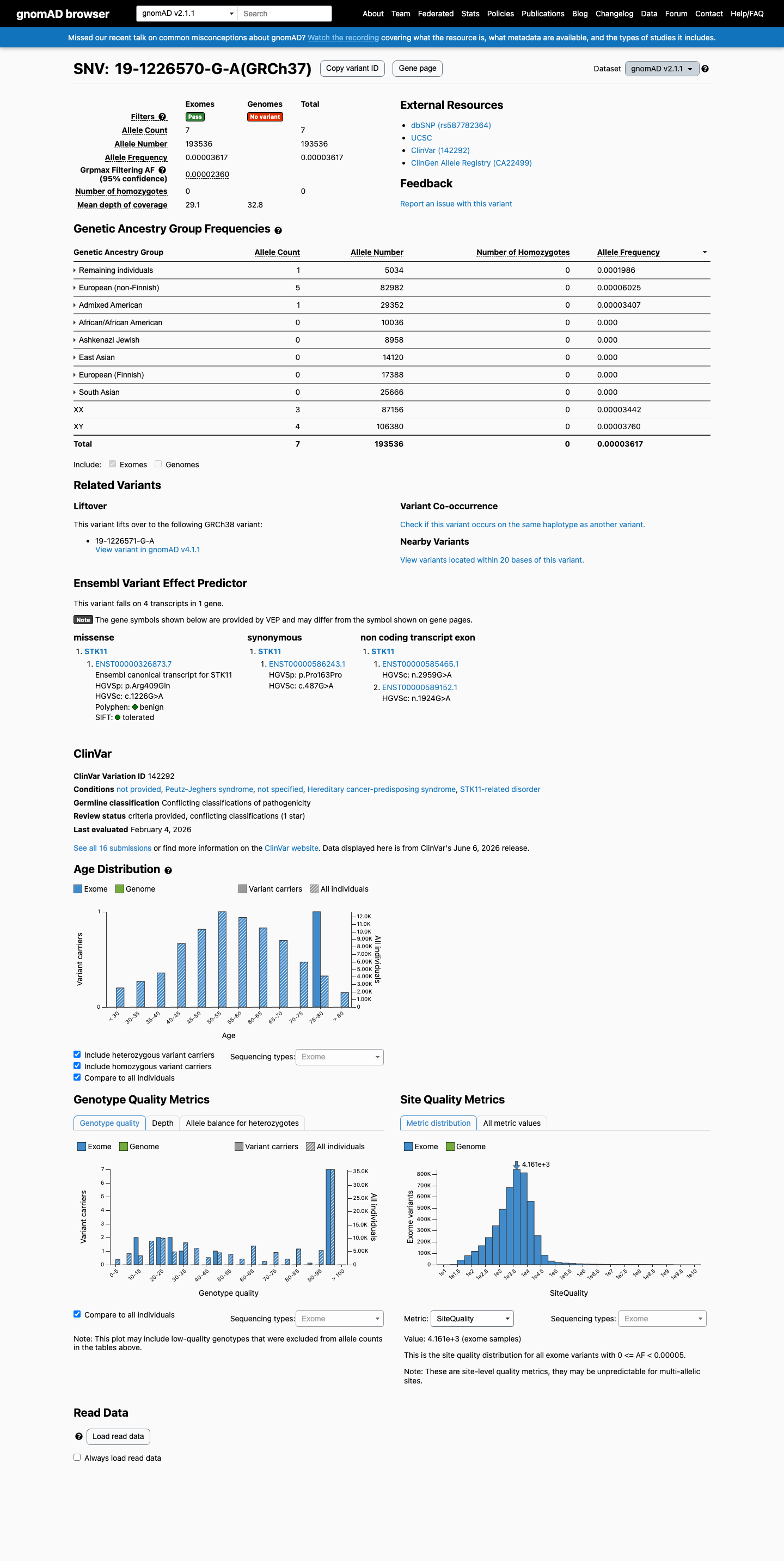
This variant is present in gnomAD v2.1 (AF= 3.6169e-05; MAF= 0.00362%, 7/193536 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.000198649; MAF= 0.01986%, 1/5034 alleles, homozygotes = 0); grpmax FAF= 2.36e-05.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0001086130118388183, 2/18414 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0033%
· 52 / 1,587,284
0 hom · FAF 0.0031%
0 hom · FAF 0.0031%
African/African American 3 / 74,264 |
0.004% |
European (non-Finnish) 47 / 1,168,440 |
0.004% |
East Asian 1 / 43,246 |
0.0023% |
Remaining individuals 1 / 61,366 |
0.0016% |
+ 6 not observed (Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0036%
· 7 / 193,536
0 hom · FAF 0.0024%
0 hom · FAF 0.0024%
Remaining individuals 1 / 5,034 |
0.02% |
European (non-Finnish) 5 / 82,982 |
0.006% |
Admixed American 1 / 29,352 |
0.0034% |
+ 5 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
0.011%
· 2 / 18,414
0 hom · FAF 0.035%
0 hom · FAF 0.035%
African/African American 2 / 1,020 |
0.2% |
+ 8 not observed (Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, European (non-Finnish), Remaining individuals, South Asian)
ClinVar
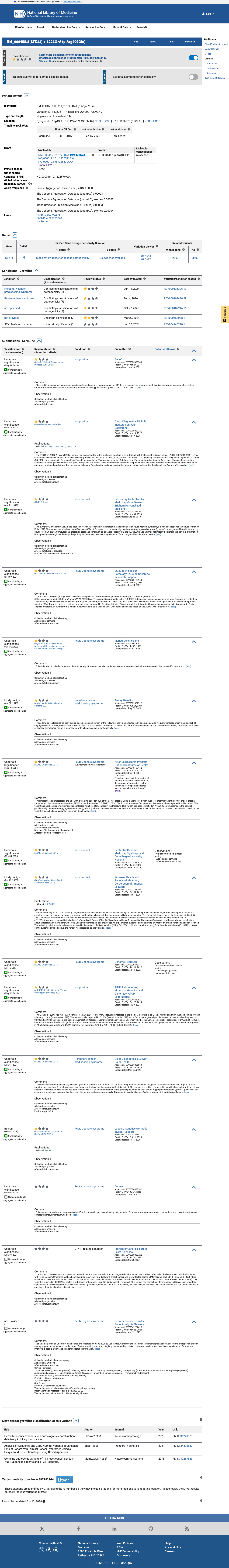
This variant has been reported in ClinVar as Uncertain significance (11 clinical laboratories) and as Likely benign (2 clinical laboratories) and as Uncertain Significance (1 clinical laboratory) and as Benign (1 clinical laboratory). (ClinVarID = 142292)
In silico
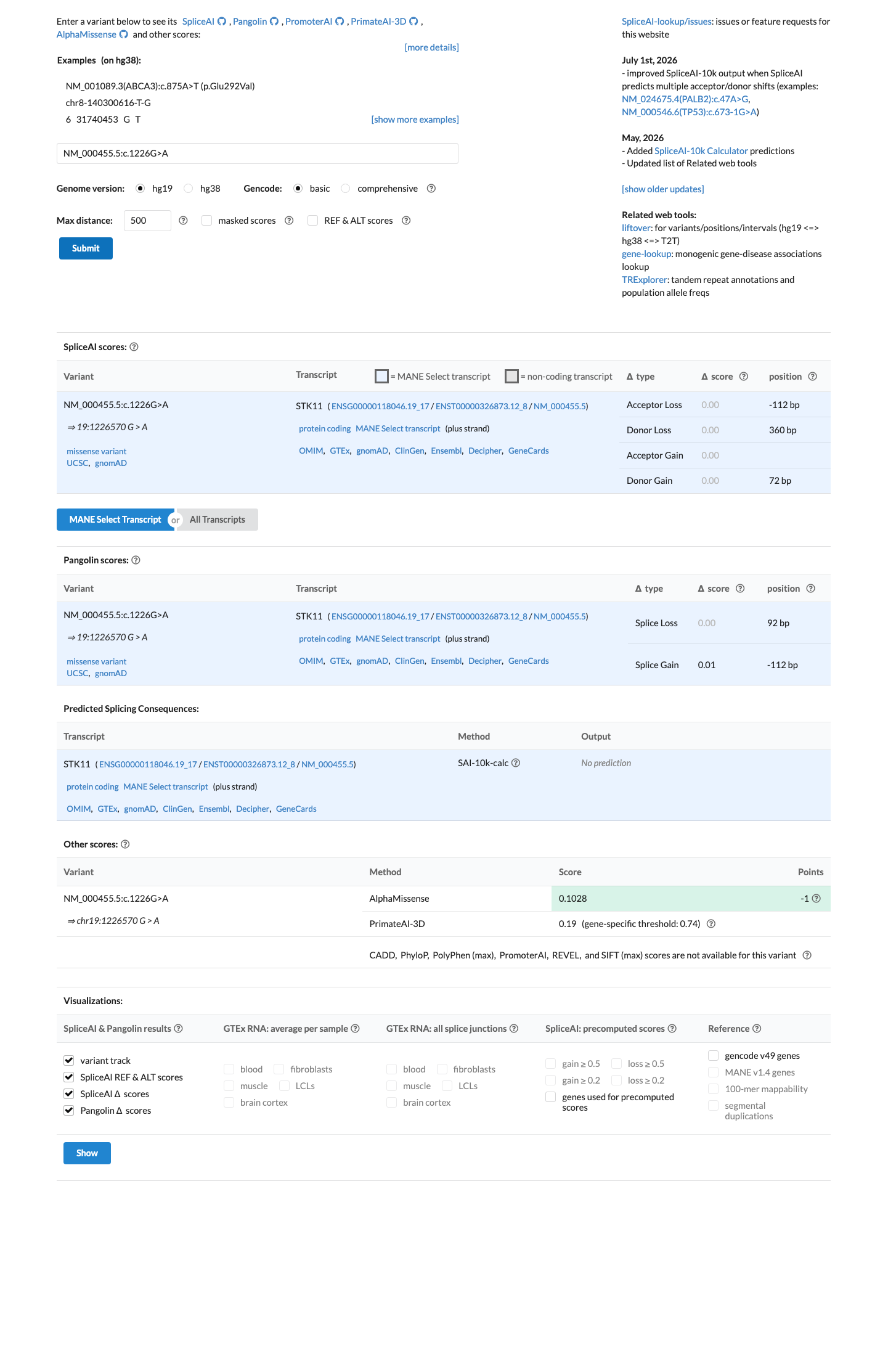
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.122. BayesDel score = -0.513561.
Functional
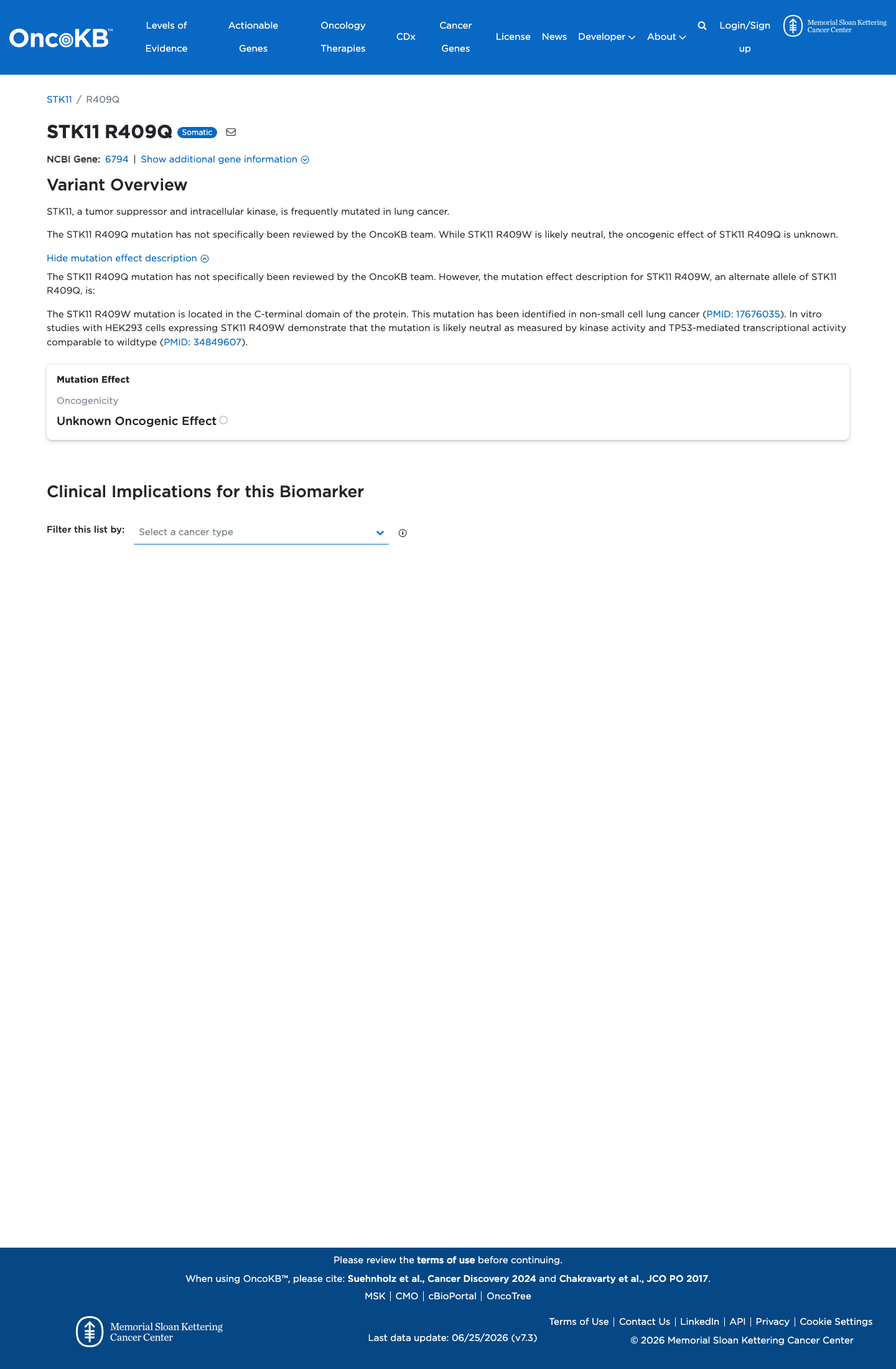
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. STK11, a tumor suppressor and intracellular kinase, is frequently mutated in lung cancer.
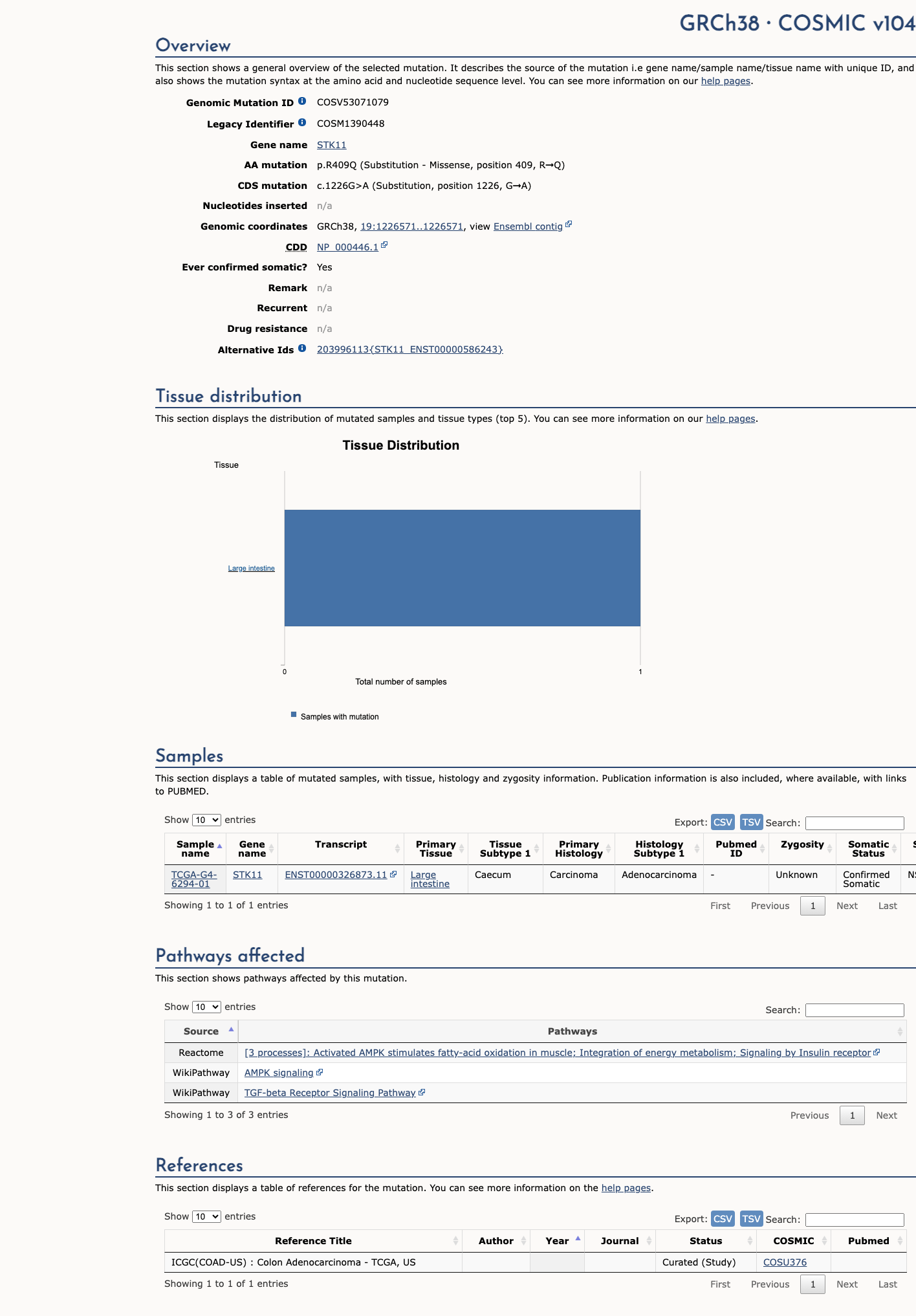
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV53071079, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
34849607 ↗
Functional assessment of somatic STK11 variants identified in primary human non-small cell lung cancers.
ONCOKB
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25645574 ↗
ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
34326862 ↗
Analysis of Sequence and Copy Number Variants in Canadian Patient Cohort With Familial Cancer Syndromes Using a Unique Next Generation Sequencing Based Approach.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR