c.7927+13T>A is present at extremely low frequency in gnomAD v4.1 (AF = 0.00075%, 12/1,609,596 alleles, 0 homozygotes; grpmax FAF = 5.01e-06), meeting the ATM HBOP VCEP threshold for PM2_Supporting (≤0.001%).1 SpliceAI predicts no significant splicing impact (max delta score = 0.03), meeting BP4_Supporting per the ATM HBOP VCEP threshold of ≤0.1.2 The variant is at the donor +13 position, beyond the +7 deep intronic boundary, meeting BP7_Supporting per the ATM HBOP VCEP as a deep intronic variant unlikely to affect splicing.3 Two lines of benign supporting evidence (BP4, BP7) are present. Applying ACMG/AMP 2015 combination rules (Richards et al. 2015, PMID:25741868), two benign supporting criteria meet the threshold for Likely Benign (Rule 19).4
ATM
Final classification
Likely Benign
ATM c.7927+13T>A · p.?
ATM
c.7927+13T>A is present at extremely low frequency in gnomAD v4.1 (AF = 0.00075%, 12/1,609,596 alleles, 0 homozygotes; grpmax FAF = 5.01e-06), meeting the ATM HBOP VCEP threshold for PM2_Supporting (≤0.001%).
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework: matched Rule19 (Benign.Supporting >=2) with applied criteria: PM2 supporting, BP4 supporting, BP7 supporting; maps to Likely Benign.
Classification rationale
PM2
BP4BP7
Likely Benign
ATM c.7927+13T>A
PM2 + BP4 + BP7
→
Likely Benign
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 10 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
c.7927+13T>A is present at extremely low frequency in gnomAD v4.1 (AF = 0.00075%, 12/1,609,596 alleles, 0 homozygotes; grpmax FAF = 5.01e-06). This frequency is ≤0.001%, meeting the ATM HBOP VCEP threshold for PM2_Supporting.
gnomAD v4.1 AF = 7.46e-06 (0.00075%)12/1609
✓
BP4
supporting
Benign
SpliceAI predicts no splicing impact for c.7927+13T>A (max delta score = 0.03). This is ≤0.1, meeting the ATM HBOP VCEP BP4 threshold for no predicted splicing impact.
SpliceAI max delta = 0.03 (≤0.1 threshold)
✓
BP7
supporting
Benign
c.7927+13T>A is an intronic variant at the donor +13 position, which is further than (but not including) +7. This meets the ATM HBOP VCEP BP7 definition for deep intronic variants unlikely to affect splicing.
Variant located at donor +13beyond the +7 deep intronic threshold
Assessed · not applied
Pathogenic
PVS1
NM_000051.4:c.7927+13T>A is an intronic substitution at the donor +13 position, outside the canonical ±1,2 splice consensus.
PS1
No comparator pathogenic or likely pathogenic splicing variant with a similar predicted splicing event has been identified for c.7927+13T>A.
PS3
No well-established in vitro or in vivo functional studies demonstrating a damaging effect on ATM protein function or splicing have been identified for c.7927+13T>A.
PS4
No case-control studies demonstrating significant enrichment of c.7927+13T>A in affected individuals versus controls have been identified.
PP1
No segregation data are available for c.7927+13T>A.
PP3
SpliceAI predicts no significant splicing impact (max delta score = 0.03).
Benign
BA1
gnomAD v4.1 grpmax filtering allele frequency is 5.01e-06 (0.000501%), which is not >0.5%.
BS1
gnomAD v4.1 grpmax filtering allele frequency is 5.01e-06 (0.000501%), which is not >0.05%.
BS3
No well-established in vitro or in vivo functional studies demonstrating no damaging effect on ATM protein function or splicing have been identified for c.7927+13T>A.
BP2
No data are available regarding observation of c.7927+13T>A in trans with a pathogenic ATM variant in unaffected individuals.
N/A · 12
PS2 · PM1 · PM5 · PM6 · PP2 · PP4 · PP5 · BS2 · BS4 · BP1 · BP5 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
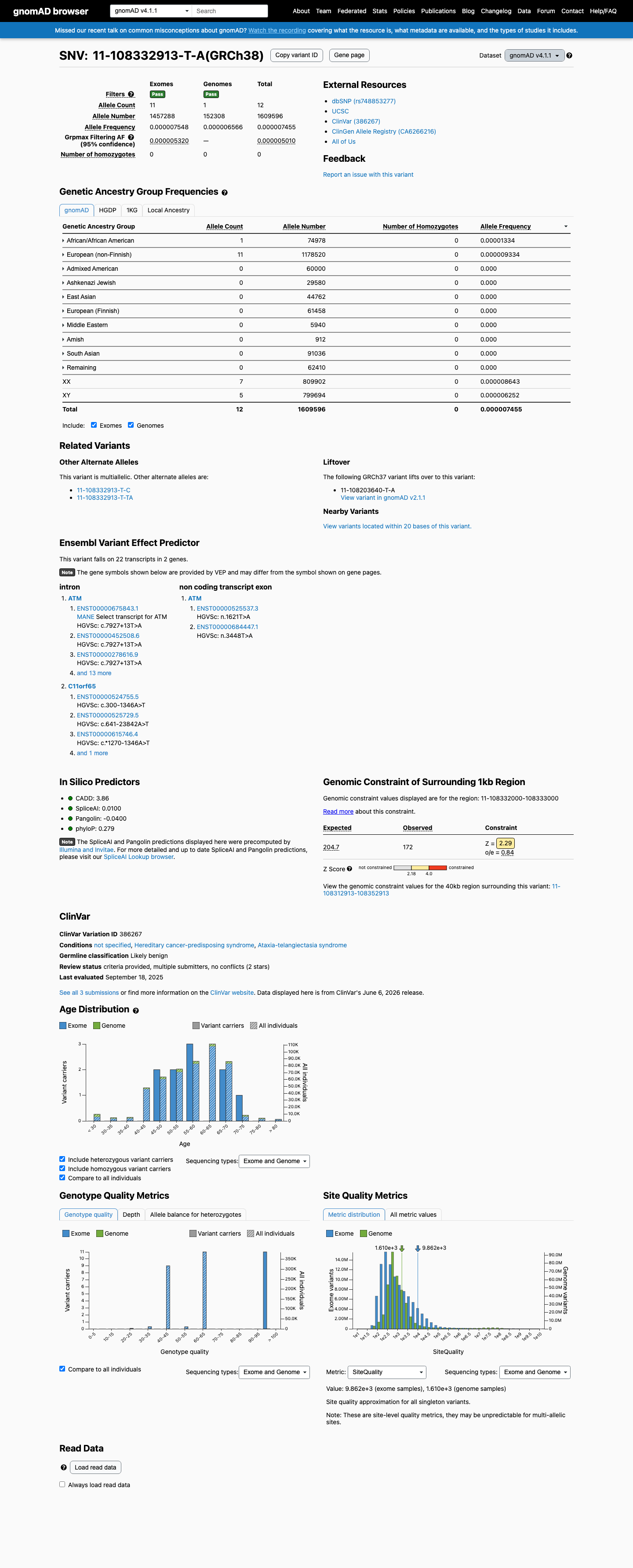
This variant is present in gnomAD v4.1 (AF= 7.45529e-06; MAF= 0.00075%, 12/1609596 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 1.33372e-05; MAF= 0.00133%, 1/74978 alleles, homozygotes = 0); grpmax FAF= 5.01e-06.
v2.1
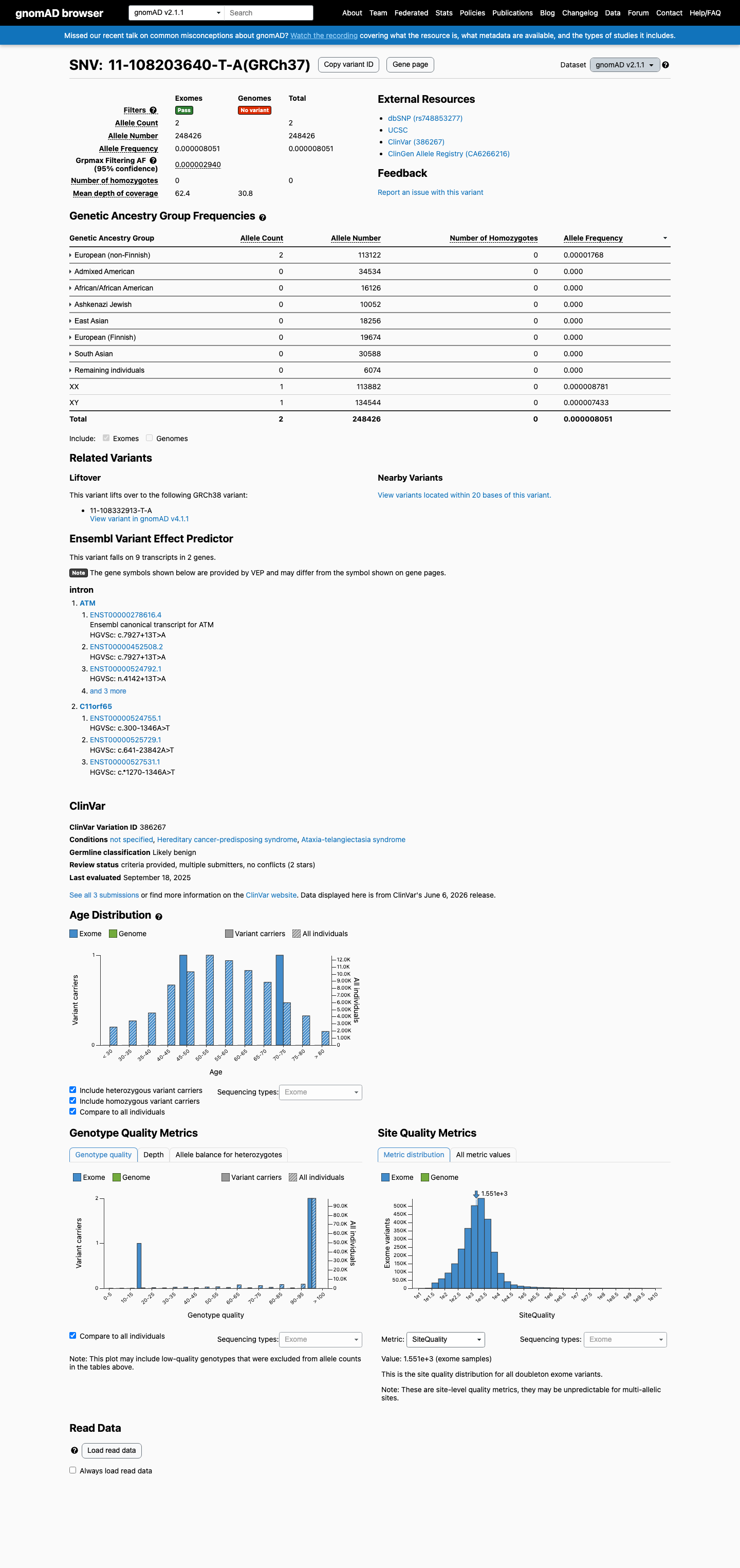
This variant is present in gnomAD v2.1 (AF= 8.05069e-06; MAF= 0.00081%, 2/248426 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.768e-05; MAF= 0.00177%, 2/113122 alleles, homozygotes = 0); grpmax FAF= 2.94e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00075%
· 12 / 1,609,596
0 hom · FAF 0.0005%
0 hom · FAF 0.0005%
African/African American 1 / 74,978 |
0.0013% |
European (non-Finnish) 11 / 1,178,520 |
0.00093% |
+ 8 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.00081%
· 2 / 248,426
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
European (non-Finnish) 2 / 113,122 |
0.0018% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
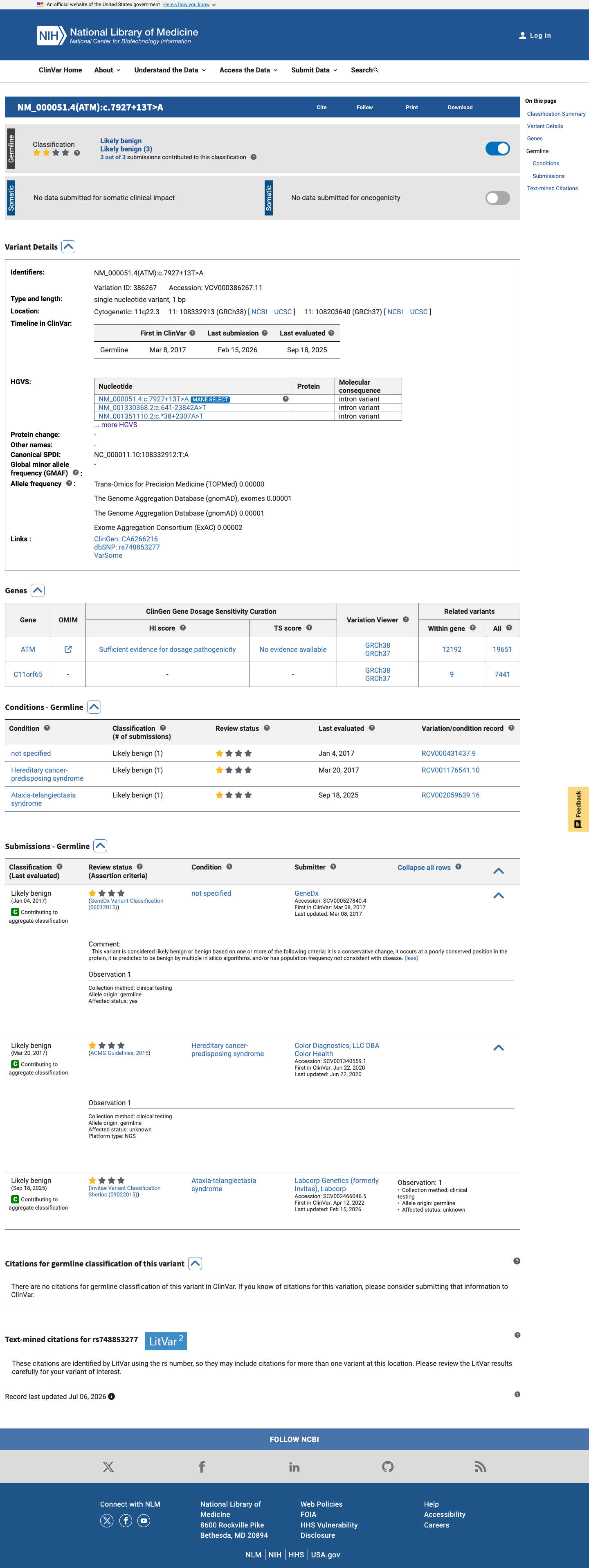
This variant is present in ClinVar (Variation ID: 386267); submission details unavailable.
In silico
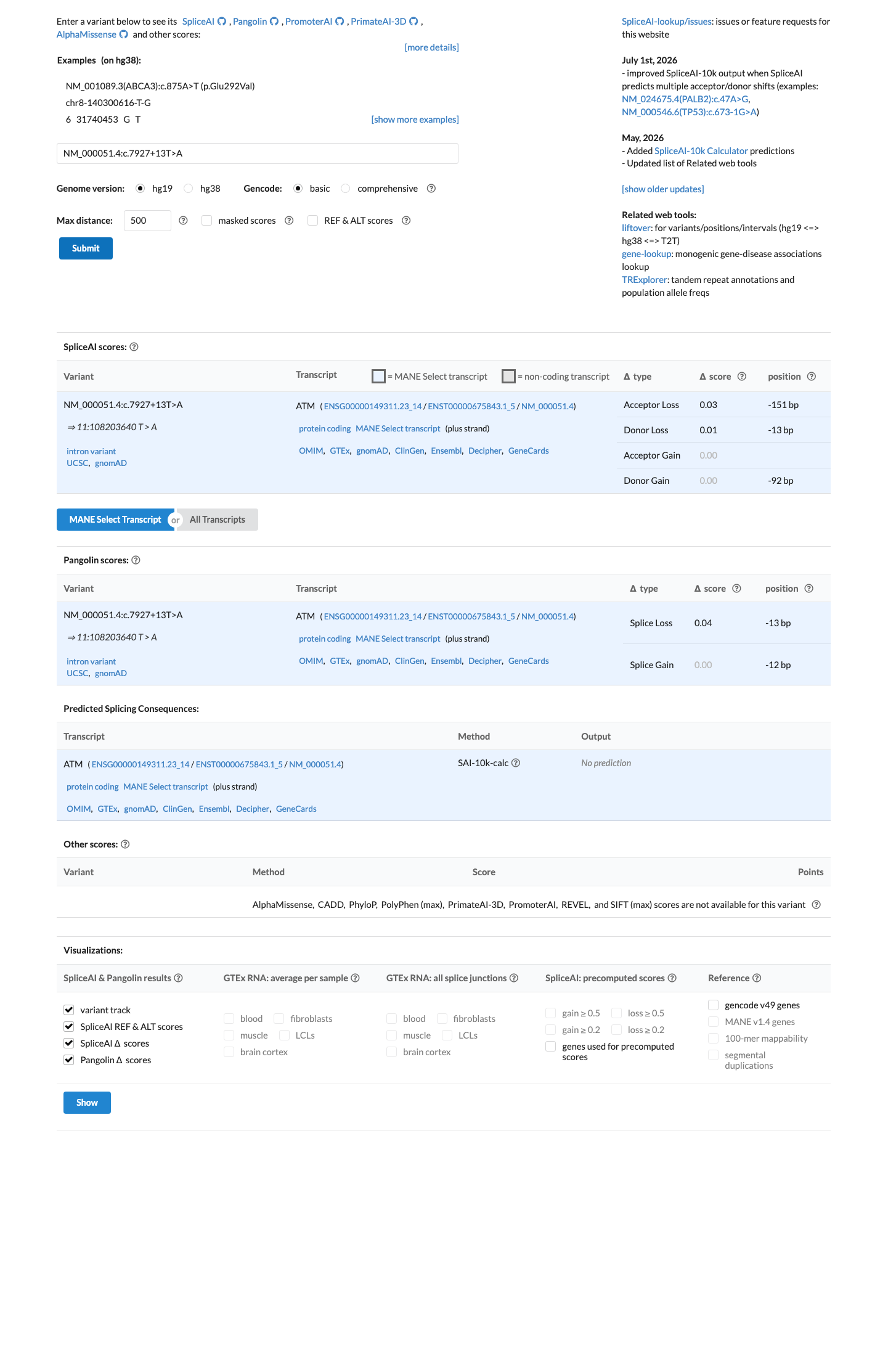
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
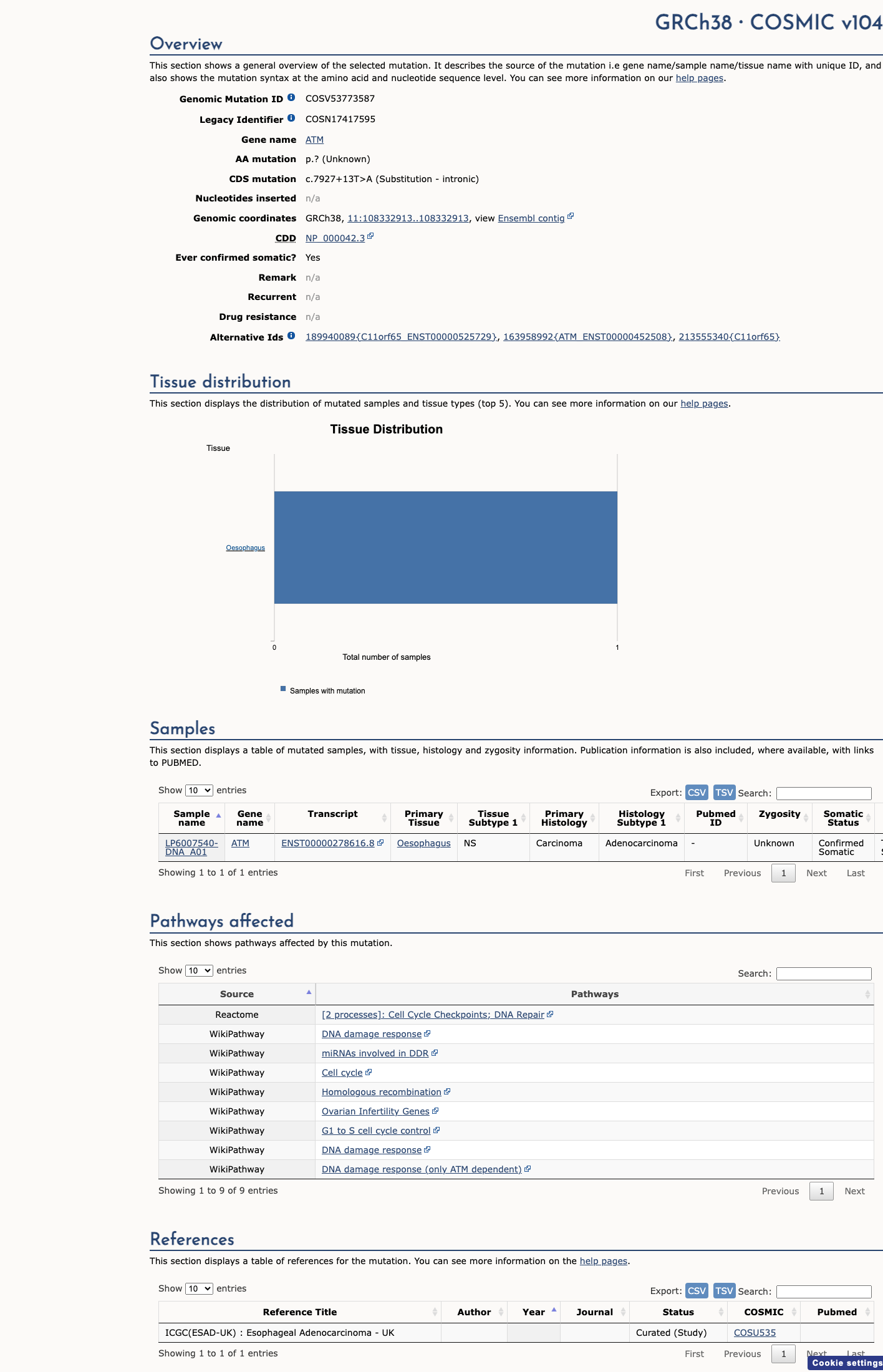
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV53773587, n = 1 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
24418350 ↗
EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR