BP1_Strong is met: c.1501A>G (p.Ile501Val) is a missense variant located at amino acid 501, outside both BRCA2 clinically important functional domains (PALB2 binding aa 10-40; DNA binding aa 2481-3186), with no predicted splicing impact (SpliceAI max delta 0.0).1 PM2_Supporting is met: the variant is absent from gnomAD v2.1 (non-cancer, exome) and observed as a single allele in gnomAD v4.1 (AF=6.27e-07), which ENIGMA deems not informative for an outbred population.2 No pathogenic criteria beyond PM2_Supporting were met. PVS1, PS1, PS3, PS4, PP1, PP3, and PP4 are all not met or not applicable for this missense variant.3 In silico predictions are consistent with a benign interpretation: BayesDel no-AF score -0.472575 and REVEL 0.228 are below pathogenic thresholds. The variant is not in a functional domain, not at a hotspot, and was not identified in any calibrated functional assay (ENIGMA Table 9).4 ClinVar reports this variant as Uncertain Significance (4 submitters) and Likely Benign (1 submitter), with no expert panel classification (ClinVar VariationID: 433760).5
BRCA2
Final classification
Likely Benign
BRCA2 c.1501A>G · p.Ile501Val
BRCA2
BP1_Strong is met: c.1501A>G (p.Ile501Val) is a missense variant located at amino acid 501, outside both BRCA2 clinically important functional domains (PALB2 binding aa 10-40; DNA binding aa 2481-3186), with no predicted splicing impact (SpliceAI max delta 0.0).
ENIGMA BRCA2 VCEP v1.2.0 conflicting-evidence point system: PM2_Supporting (+1 pathogenic point) and BP1_Strong (-4 benign point) = -3 total points, which falls in the Likely Benign range (-6 to -2). Both pathogenic and benign criteria are met, triggering the ENIGMA Table 3 point-based conflicting evidence rule.
Classification rationale
PM2
BP1
Likely Benign
BRCA2 c.1501A>G
PM2 + BP1
→
Likely Benign
3
cspec ↗
4
bayesdelrevelvcep_specifications_table9_v1_2_2024_11_18
Gene diagram
· NM_000059.4 · variants mapped to exon structure
BRCA2
NM_000059.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 14 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
review
Pathogenic
The variant is absent from gnomAD v2.1 (non-cancer, exome). In gnomAD v4.1, it is observed as a single allele (AF=6.27e-07, 1/1,595,636) in the European (non-Finnish) population. Per ENIGMA guidance, a single observation in a gnomAD outbred population is not informative and does not preclude PM2_Supporting. The variant is also absent from gnomAD-Canada.
Absent from gnomAD v2.1 (non-cancerexome only).Single allele in gnomAD v4.1 (AF=6.27e-07
✓
BP1
strong
Benign
ENIGMA BP1_Strong applies to missense variants outside a (potentially) clinically important functional domain with no predicted splicing impact (SpliceAI ≤ 0.1). This missense variant (p.Ile501Val) is at amino acid position 501, which is outside both BRCA2 clinically important functional domains (PALB2 binding: aa 10-40; DNA binding: aa 2481-3186). SpliceAI max delta is 0.0, indicating no splicing impact. BRCA2 exons 10 and 11 (codons 266-2281) contain 0 P/LP missense variants among 2,177 submitted to ClinVar, consistent with this region being a validated coldspot.
Missense variant at position 501outside PALB2 binding domain (aa 10-40) and DNA binding domain (aa 2481-3186).SpliceAI max delta 0.0 — no splicing impact.
Assessed · not applied
Pathogenic
PS1
No previously classified pathogenic or likely pathogenic variant with the same amino acid change (p.Ile501Val) was identified.
PS3
This variant was not identified in the ENIGMA Specifications Table 9 curated functional assay results.
PS4
No case-control study demonstrating significantly increased prevalence in affected individuals was identified.
PP1
No co-segregation data or quantitative likelihood ratio analysis is available for this variant.
PP3
ENIGMA PP3 requires either (a) a missense variant inside a clinically important functional domain with BayesDel no-AF ≥ 0.30, or (b) SpliceAI ≥ 0.2.
PP4
ENIGMA PP4 requires a combined likelihood ratio from multifactorial clinical data (LR ≥ 2.08 for Supporting).
Benign
BA1
ENIGMA BA1 requires FAF > 0.1% (0.001) in gnomAD non-founder populations.
BS1
ENIGMA BS1_Supporting requires FAF > 0.002% (0.00002) and BS1_Strong requires FAF > 0.01% (0.0001).
BS2
ENIGMA BS2 requires co-occurrence of the variant in trans with a known P/LP variant in the same gene, with absence of Fanconi Anemia phenotype, scored per Specifications Table 8.
BS3
The variant was not identified in the ENIGMA Specifications Table 9 curated functional assay results assigning BS3 codes.
BS4
ENIGMA BS4 requires lack of segregation in affected family members as measured by quantitative co-segregation analysis (LR ≤ 0.48 for Supporting).
BP4
ENIGMA BP4_Supporting applies to missense variants inside a clinically important functional domain with BayesDel no-AF ≤ 0.18 and SpliceAI ≤ 0.1.
BP5
ENIGMA BP5 requires a combined likelihood ratio against pathogenicity from multifactorial clinical data (LR ≤ 0.48 for Supporting).
BP7
ENIGMA BP7_Strong (RNA) requires well-established mRNA functional studies showing no damaging effect.
N/A · 9
PVS1 · PS2 · PM1 · PM5 · PM6 · PP2 · PP5 · BP2 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
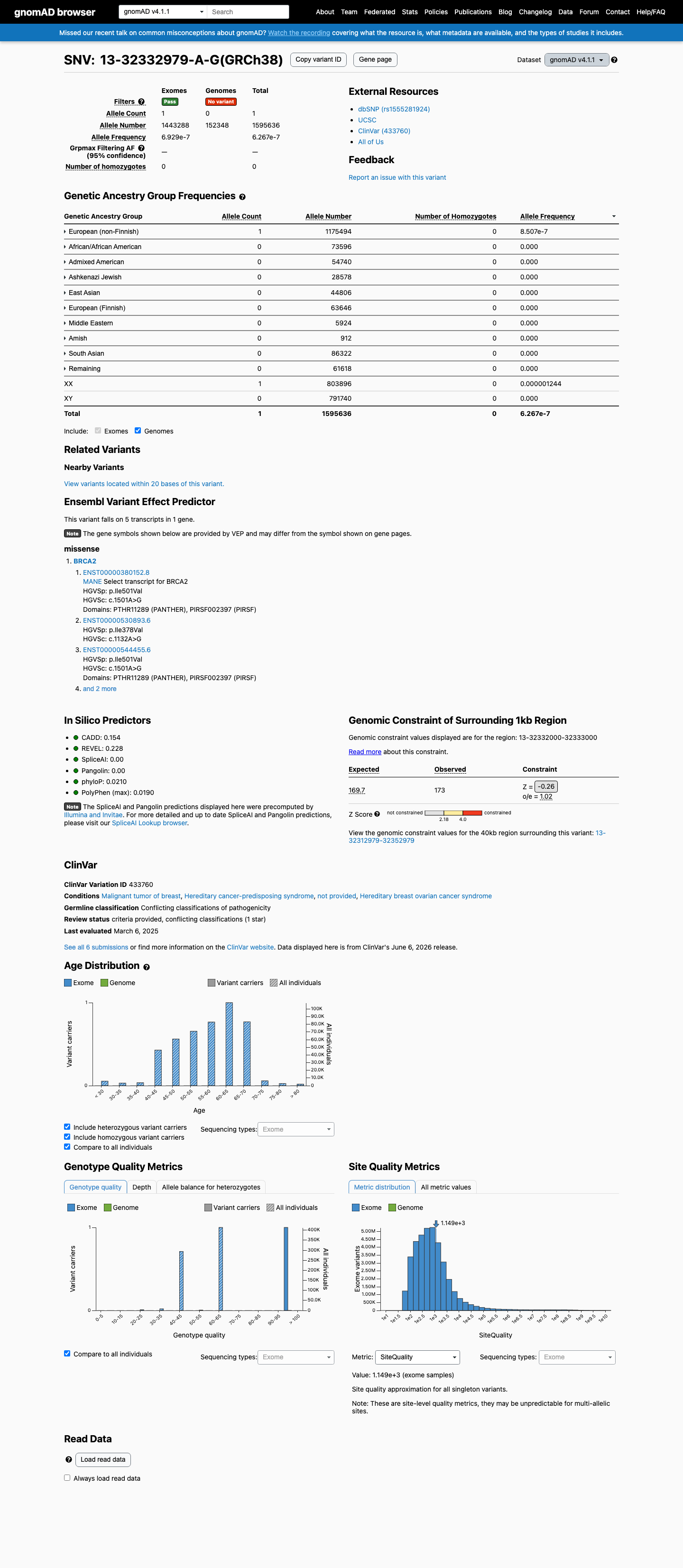
This variant is present in gnomAD v4.1 (AF= 6.26709e-07; MAF= 0.00006%, 1/1595636 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.50706e-07; MAF= 0.00009%, 1/1175494 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.3e-05%
· 1 / 1,595,636
0 hom
0 hom
European (non-Finnish) 1 / 1,175,494 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
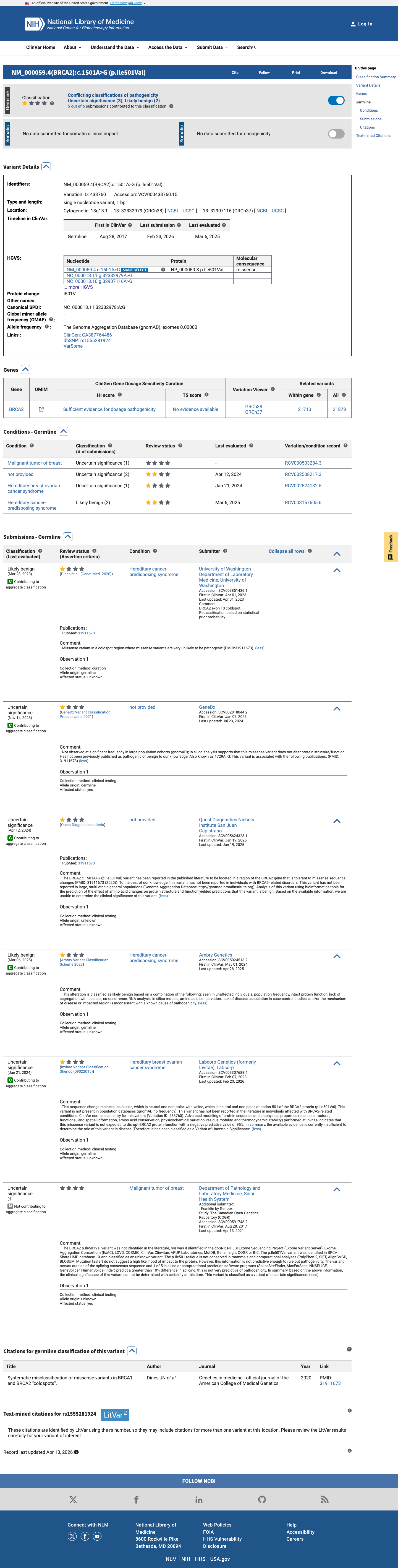
This variant has been reported in ClinVar as Uncertain significance (4 clinical laboratories) and as Likely benign (1 clinical laboratory). (ClinVarID = 433760)
In silico
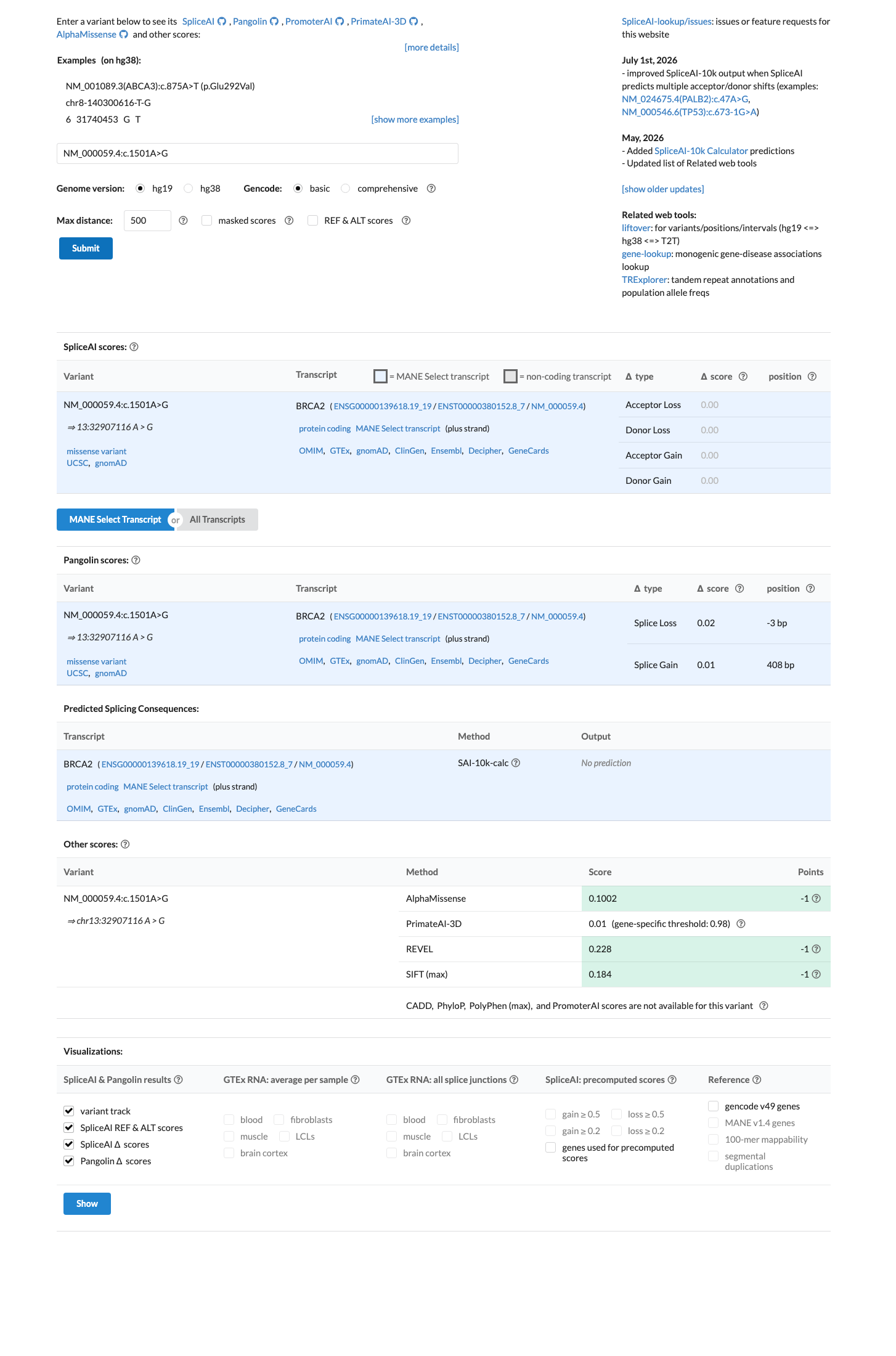
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.228. BayesDel score = -0.472575.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. BRCA2, a tumor suppressor involved in the DNA damage response, is mutated in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Systematic misclassification of missense variants in BRCA1 and BRCA2 "coldspots".
Searched
c.1501A>Gp.Ile501ValI501VIle501Val1501
Found
Evaluated BRCA1 and BRCA2 missense variants in ClinVar to identify coldspot regions. In BRCA2 exons 10 and 11 (codons 266-2281), 0 of 2,177 missense variants were classified as pathogenic or likely pathogenic, establishing this region as a coldspot. Proposed that missense variants in coldspots can be classified as likely benign using Bayesian odds ratio thresholds.
Variant
◇ Residue / gene-level — variant not named
Applied to
→BP1 supports · met
Why
Variant not specifically mentioned, but paper provides foundational coldspot evidence supporting BP1_Strong application for missense variants in BRCA2 exon 10 (codon 501). 0/2177 P/LP missense variants in this region.
Exon 10 and 11 ... 0 (0) ... 110 (5.1) ... 2067 (94.9) ... 2177 (100.0)
Location Table 1; Results, BRCA2 section · full text
Sources & reference links
9Sources
Triaged references · 6 PMIDs not cited in assessment
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
12692171 ↗
American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility.
CLINVAR
20065170 ↗
American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility.
CLINVAR
23188549 ↗
NSGC practice guideline: risk assessment and genetic counseling for hereditary breast and ovarian cancer.
CLINVAR
24493721 ↗
American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers.
CLINVAR