NM_000535.7:c.705+24C>T is an intronic variant in PMS2 located at position +24 of intron 6, outside the canonical splice consensus region.1 This variant is present in gnomAD v4.1 at an extremely low overall allele frequency of 1.18e-05 (19/1,610,524 alleles, no homozygotes), meeting PM2_Supporting per InSiGHT PMS2 VCEP criteria (allele frequency <0.00002).2 SpliceAI predicts no splicing impact with a maximum delta score of 0.04, meeting BP4_Supporting per the InSiGHT PMS2 VCEP (delta ≤0.1 for intronic variants).3 The variant is at intronic position +24, which is beyond the +7 canonical splice donor boundary, meeting BP7_Supporting per the InSiGHT PMS2 VCEP (intronic variants at or beyond -21/+7).4 The variant is absent from ClinVar and no publications were identified that mention NM_000535.7:c.705+24C>T.5 The variant has been observed in COSMIC (COSV113734575, n=2 somatic occurrences), but this does not constitute germline evidence of pathogenicity. Criteria met: PM2_Supporting (extremely rare in population databases). Benign criteria met: BP4_Supporting (SpliceAI predicts no splice impact), BP7_Supporting (intronic variant beyond canonical splice region).6 Per InSiGHT PMS2 VCEP v2.0.0 combination Rule 31, the co-occurrence of pathogenic supporting evidence (PM2) and benign supporting evidence (BP4, BP7) results in Uncertain Significance — Conflicting Evidence. While the variant also satisfies Rule 19 (≥2 benign supporting criteria for Likely Benign), the presence of pathogenic supporting evidence triggers the conflicting evidence rule.7
PMS2
Final classification
Likely Benign
PMS2 c.705+24C>T · p.?
PMS2
NM_000535.7:c.705+24C>T is an intronic variant in PMS2 located at position +24 of intron 6, outside the canonical splice consensus region.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PMS2 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule19 (Benign.Supporting >=2) with applied criteria: PM2 supporting, BP4 supporting benign, BP7 supporting benign; maps to Likely Benign.
Classification rationale
PM2
BP4BP7
Likely Benign
PMS2 c.705+24C>T
PM2 + BP4 + BP7
→
Likely Benign
Gene diagram
· NM_000535.7 · variants mapped to exon structure
PMS2
NM_000535.7
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 12 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000535.7:c.705+24C>T has an overall allele frequency of 1.18e-05 (19/1,610,524 alleles) in gnomAD v4.1, which is below the VCEP PM2 threshold of <0.00002 (<1 in 50,000 alleles). The variant is absent from gnomAD-Canada v1.0. Per the InSiGHT PMS2 VCEP v2.0.0, this meets PM2_Supporting.
gnomAD v4.1 total AF = 1.18e-05 (19/1610524 alleles)
✓
BP4
supporting
Benign
NM_000535.7:c.705+24C>T is an intronic variant. SpliceAI predicts no splicing impact with a maximum delta score of 0.04, which is ≤0.1. Per the InSiGHT PMS2 VCEP v2.0.0, intronic variants with SpliceAI delta ≤0.1 meet BP4_Supporting (Walker et al. 2023).
SpliceAI max delta = 0.04below BP4 threshold of ≤0.1 for intronic variants.
✓
BP7
supporting
Benign
NM_000535.7:c.705+24C>T is an intronic variant located at position +24 in intron 6 of PMS2, which is beyond the +7 canonical splice donor boundary. Per the InSiGHT PMS2 VCEP v2.0.0, intronic variants at or beyond -21/+7 meet BP7_Supporting. This criterion may be combined with BP4 as noted in the VCEP rule.
Intronic variant at position +24beyond the +7 canonical splice donor boundary.
Assessed · not applied
Pathogenic
PS1
PS1 requires either a predicted missense substitution encoding the same amino acid change as a previously established pathogenic variant, or a variant affecting the same non-canonical splice nucleotide as a confirmed pathogenic splice variant with similar or worse SpliceAI prediction.
PS2
No de novo observations are available for NM_000535.7:c.705+24C>T.
PS3
No functional assay data are available for NM_000535.7:c.705+24C>T.
PP1
No co-segregation data are available for NM_000535.7:c.705+24C>T.
PP3
PP3 in the InSiGHT PMS2 VCEP requires either an HCI prior score >0.68 for missense variants or a SpliceAI delta score ≥0.2 for non-canonical splice variants.
PP4
No tumor phenotype data are available for NM_000535.7:c.705+24C>T.
Benign
BA1
The gnomAD v4.1 grpmax filtering allele frequency for NM_000535.7:c.705+24C>T is 3.59e-05 (0.00359%), which is far below the VCEP BA1 threshold of ≥0.0028 (0.28%).
BS1
The gnomAD v4.1 grpmax filtering allele frequency for NM_000535.7:c.705+24C>T is 3.59e-05 (0.00359%), which is below the VCEP BS1 threshold range of ≥0.00028 and <0.0028 (0.028%-0.28%).
BS2
BS2 requires co-occurrence in trans with a known pathogenic PMS2 variant in a patient with colorectal cancer after age 45 without CMMRD features.
BS3
BS3 requires calibrated functional assays showing benign functional odds (≤0.05 for Strong, >0.05 & ≤0.48 for Supporting), or for intronic variants, laboratory assays demonstrating no associated mRNA aberration with NMD inhibition.
BS4
No co-segregation data are available for NM_000535.7:c.705+24C>T.
BP5
No tumor phenotype data are available for NM_000535.7:c.705+24C>T.
N/A · 13
PVS1 · PS4 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
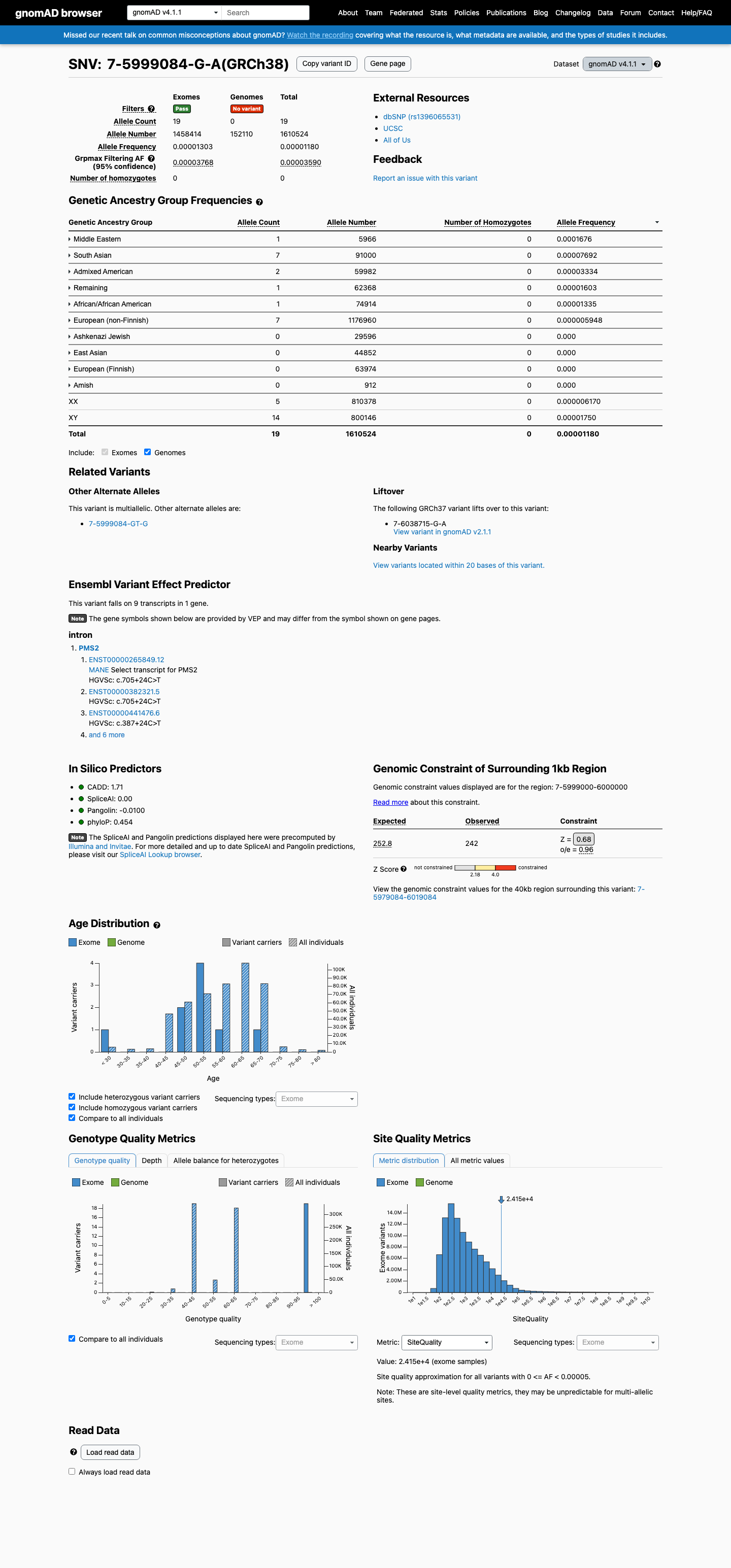
This variant is present in gnomAD v4.1 (AF= 1.17974e-05; MAF= 0.00118%, 19/1610524 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000167616; MAF= 0.01676%, 1/5966 alleles, homozygotes = 0); grpmax FAF= 3.59e-05.
v2.1
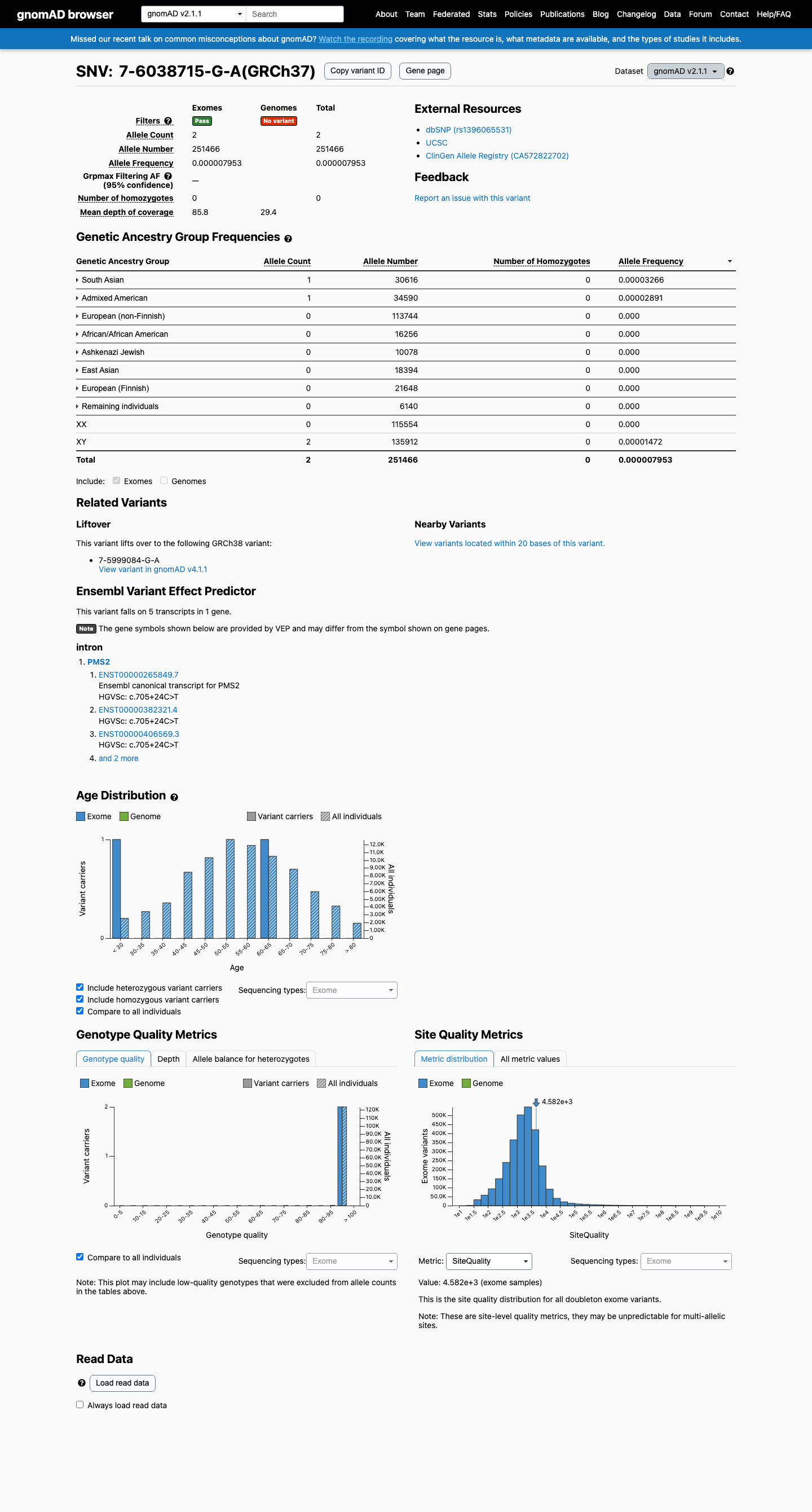
This variant is present in gnomAD v2.1 (AF= 7.95336e-06; MAF= 0.00080%, 2/251466 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 3.26627e-05; MAF= 0.00327%, 1/30616 alleles, homozygotes = 0).
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0012%
· 19 / 1,610,524
0 hom · FAF 0.0036%
0 hom · FAF 0.0036%
Middle Eastern 1 / 5,966 |
0.017% |
South Asian 7 / 91,000 |
0.0077% |
Admixed American 2 / 59,982 |
0.0033% |
Remaining individuals 1 / 62,368 |
0.0016% |
African/African American 1 / 74,914 |
0.0013% |
European (non-Finnish) 7 / 1,176,960 |
0.00059% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0008%
· 2 / 251,466
0 hom
0 hom
South Asian 1 / 30,616 |
0.0033% |
Admixed American 1 / 34,590 |
0.0029% |
+ 6 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
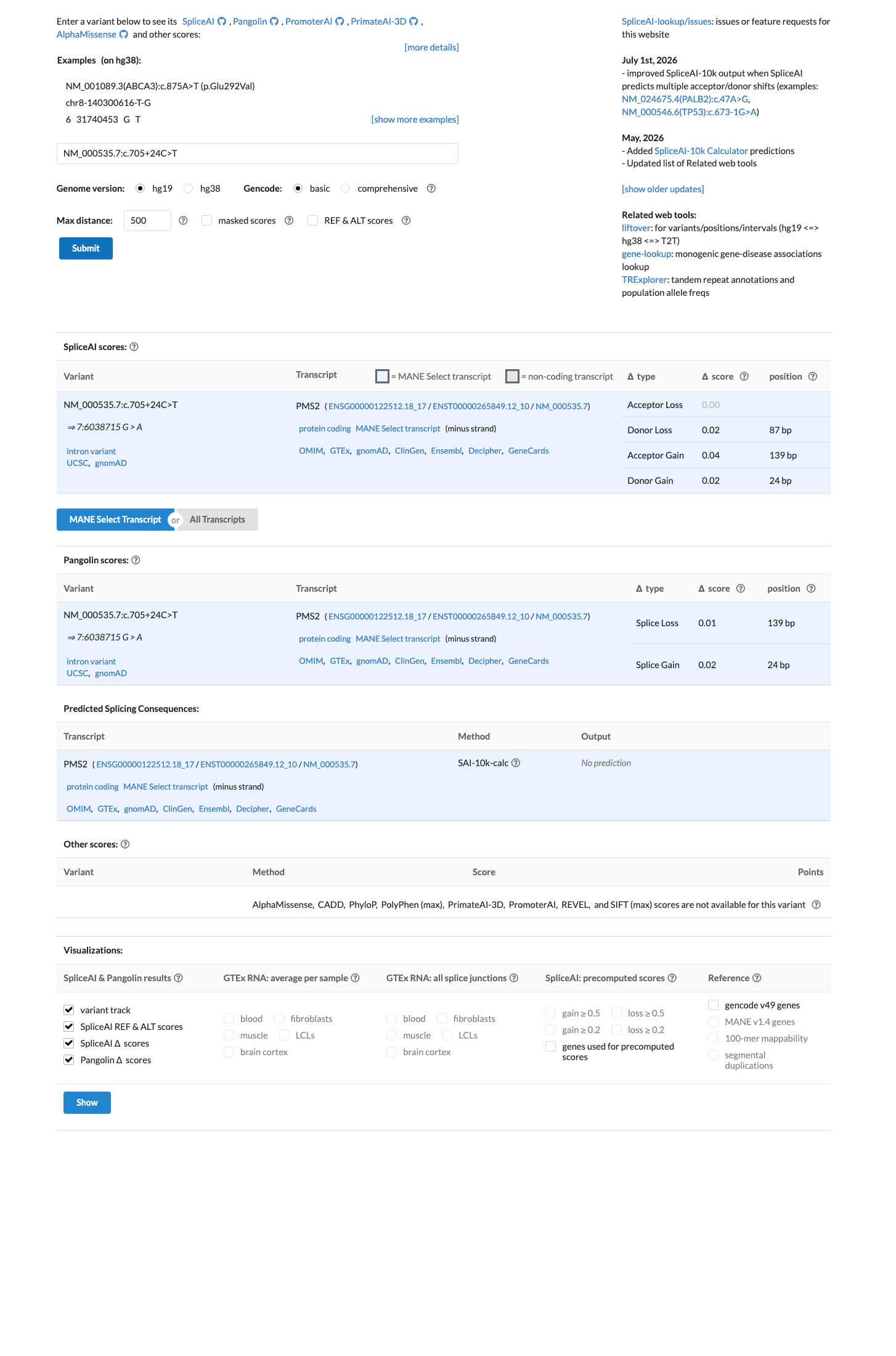
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.04).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV113734575, n = 2 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links