NM_006231.4:c.6330+19G>A is classified as Benign. BA1 is met: the variant has a grpmax filtering allele frequency of 4.01% (gnomAD v2.1) and 4.05% (gnomAD v4.1), exceeding the 1% stand-alone benign threshold. The variant is observed in 1169/280568 alleles (16 homozygotes) in gnomAD v2.1 and 3498/1600686 alleles (54 homozygotes) in gnomAD v4.1, with highest frequency in the African/African American population (4.23% v2.1, 4.17% v4.1).1 BP4 (supporting benign) is met: SpliceAI predicts no splicing impact (max delta = 0.00).2 BP6 (supporting benign) is met: ClinVar reports this variant as Benign from 6 independent clinical laboratories.3 All pathogenic criteria were assessed and are either not met, not applicable, or not supported by the available evidence. PVS1 is not applicable (deep intronic, not a null variant). PM1 is not applicable (not an exonuclease-domain missense hotspot). PM2 is not met (allele frequency exceeds 0.1%). PP3 is not met (SpliceAI delta = 0, no REVEL score available). BA1 alone satisfies the Benign classification rule under both the León-Castillo custom POLE framework and generic ACMG/AMP 2015 combination rules.4
POLE
Final classification
Benign
POLE c.6330+19G>A · p.?
POLE
NM_006231.4:c.6330+19G>A is classified as Benign.
BA1 (stand-alone benign) is met: the variant has grpmax filtering allele frequency of 4.01% (gnomAD v2.1) and 4.05% (gnomAD v4.1), exceeding the 1% stand-alone benign threshold, with 16+54 homozygotes observed. Under both the León-Castillo custom POLE framework (rule: 'BA1') and generic ACMG/AMP 2015 combination rules, BA1 alone suffices for a Benign classification. BP4 and BP6 (both supporting benign) provide additional concordant evidence but are not required for the call.
Classification rationale
BA1BP4BP6
Benign
POLE c.6330+19G>A
BA1 + BP4 + BP6
→
Benign
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 9 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
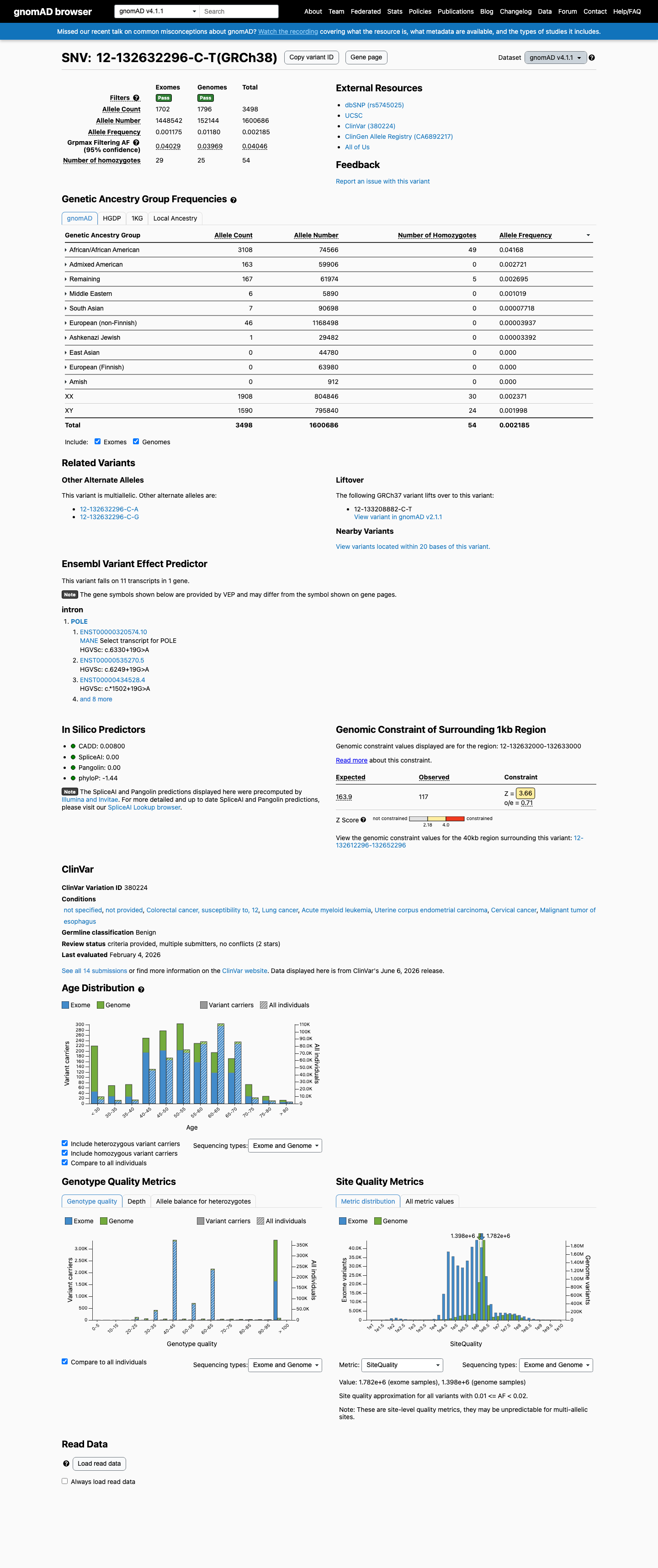
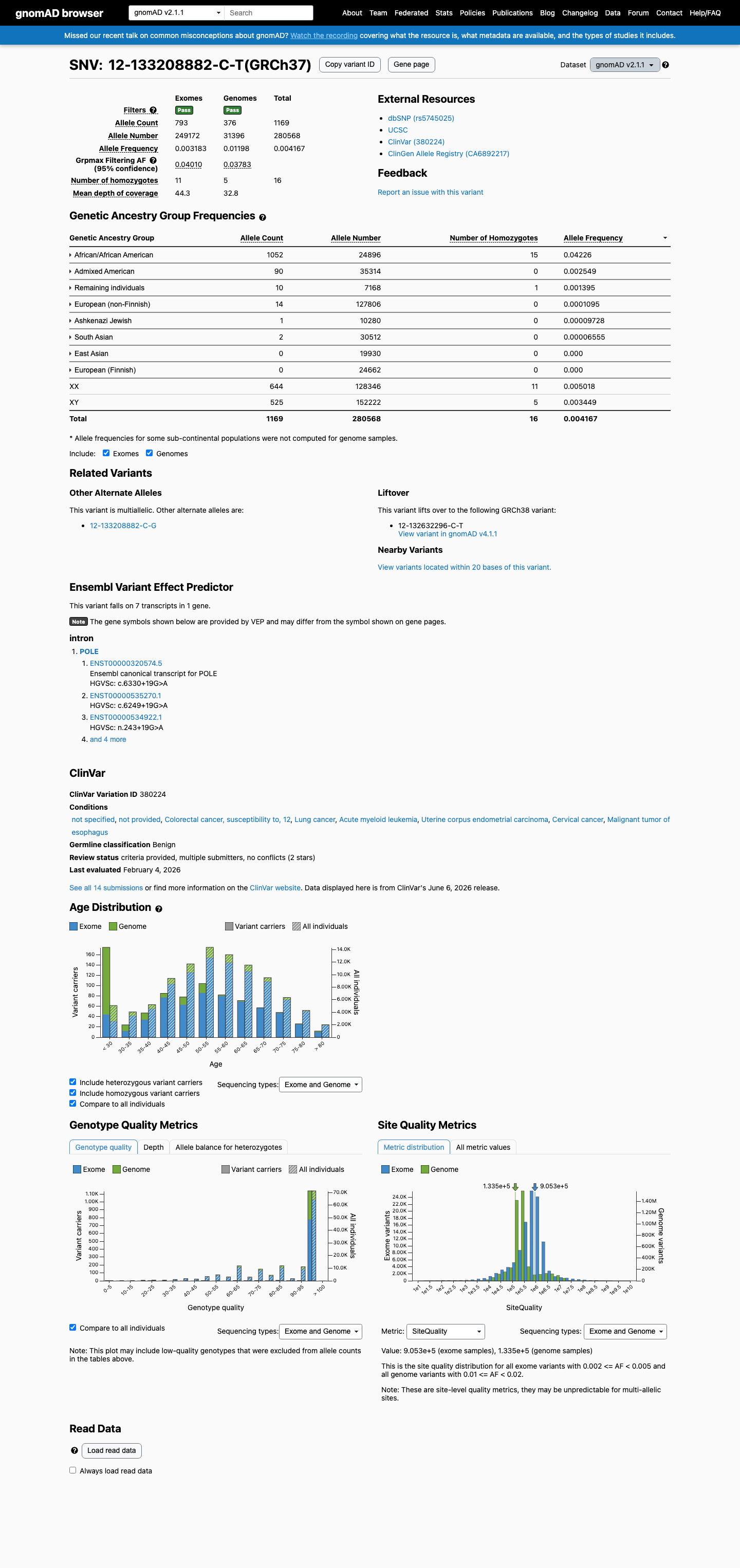
This variant is present at high frequency in gnomAD population databases. In gnomAD v2.1: overall AF=0.4166% (1169/280568 alleles), African/African American subpopulation AF=4.23% (1052/24896 alleles), grpmax FAF=4.01%, with 16 homozygotes observed. In gnomAD v4.1: overall AF=0.2185% (3498/1600686 alleles), African/African American subpopulation AF=4.17% (3108/74566 alleles), grpmax FAF=4.05%, with 54 homozygotes observed. The grpmax filtering allele frequency exceeds the BA1 threshold of >1% in both datasets, and the observation of multiple homozygotes confirms this is a common population polymorphism incompatible with a Mendelian disease-causing role.
gnomAD v2.1: overall AF=0.4166%African/African American AF=4.23%grpmax FAF=4.01%
✓
BP4
supporting
Benign
SpliceAI predicts no splicing impact for this intronic variant (max delta score = 0.00), indicating that the c.6330+19G>A substitution does not create or disrupt a splice site. In the absence of applicable missense prediction tools (REVEL and BayesDel not available for intronic variants), SpliceAI provides the primary computational evidence for this variant class, and its prediction of no splicing effect supports a benign interpretation.
SpliceAI max delta = 0.00 — no predicted donor gain/lossno acceptor gain/lossVariant is at intronic +19 position — SpliceAI is the most relevant in silico tool for this variant type
✓
BP6
supporting
Benign
ClinVar reports this variant as Benign (6 clinical laboratories: GeneDx, PreventionGenetics, ARUP Laboratories, KCCC/NGS Laboratory, Labcorp/Invitae, and Center for Genomic Medicine Rigshospitalet). Although review status is 'criteria provided, single submitter' and no expert panel review is recorded, the unanimous classification as Benign across multiple independent clinical laboratories provides supporting evidence for a benign interpretation.
ClinVar variation ID 380224: Benign6 clinical laboratories unanimously classify as BenignSubmitters include GeneDx
Assessed · not applied
Pathogenic
PS3
No functional studies have been performed on this intronic variant.
PM2
PM2 requires absence from population databases or presence at extremely low frequency (<0.1%).
PP1
No segregation data are available for this variant.
PP3
SpliceAI predicts no splicing impact (max delta score = 0.00).
PP4
No patient phenotype data specific to this variant are available.
PP5
PP5 requires a reputable source to have classified the variant as pathogenic.
Benign
BS2
BS2 requires observation of the variant in a healthy adult individual in the homozygous state, or in trans with a pathogenic variant, with full penetrance expected at an early age for a dominant disorder.
BS3
No well-established functional studies have demonstrated no deleterious effect of this variant.
BS4
No segregation data are available demonstrating lack of cosegregation with disease.
N/A · 13
PVS1 · PS1 · PS2 · PS4 · PM1 · PM5 · PM6 · PP2 · BS1 · BP1 · BP2 · BP5 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00218531; MAF= 0.21853%, 3498/1600686 alleles, homozygotes = 54) and has highest observed frequency in the African/African American population (AF= 0.0416812; MAF= 4.16812%, 3108/74566 alleles, homozygotes = 49); grpmax FAF= 0.0404589.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00416655; MAF= 0.41665%, 1169/280568 alleles, homozygotes = 16) and has highest observed frequency in the African/African American population (AF= 0.0422558; MAF= 4.22558%, 1052/24896 alleles, homozygotes = 15); grpmax FAF= 0.0401049.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0024445893089960887, 45/18408 alleles, homozygotes = 1).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.22%
· 3498 / 1,600,686
54 hom · FAF 4%
54 hom · FAF 4%
African/African American 3108 / 74,566 |
4.2% 49 hom |
Admixed American 163 / 59,906 |
0.27% |
Remaining individuals 167 / 61,974 |
0.27% 5 hom |
Middle Eastern 6 / 5,890 |
0.1% |
South Asian 7 / 90,698 |
0.0077% |
European (non-Finnish) 46 / 1,168,498 |
0.0039% |
Ashkenazi Jewish 1 / 29,482 |
0.0034% |
+ 3 not observed (European (Finnish), Amish, East Asian)
gnomAD v2.1
0.42%
· 1169 / 280,568
16 hom · FAF 4%
16 hom · FAF 4%
African/African American 1052 / 24,896 |
4.2% 15 hom |
Admixed American 90 / 35,314 |
0.25% |
Remaining individuals 10 / 7,168 |
0.14% 1 hom |
European (non-Finnish) 14 / 127,806 |
0.011% |
Ashkenazi Jewish 1 / 10,280 |
0.0097% |
South Asian 2 / 30,512 |
0.0066% |
+ 2 not observed (East Asian, European (Finnish))
gnomAD Canada 🇨🇦
0.24%
· 45 / 18,408
1 hom · FAF 3%
1 hom · FAF 3%
African/African American 41 / 1,020 |
4% 1 hom |
Remaining individuals 3 / 1,136 |
0.26% |
Latino/Admixed American 1 / 836 |
0.12% |
+ 6 not observed (Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, European (non-Finnish), South Asian)
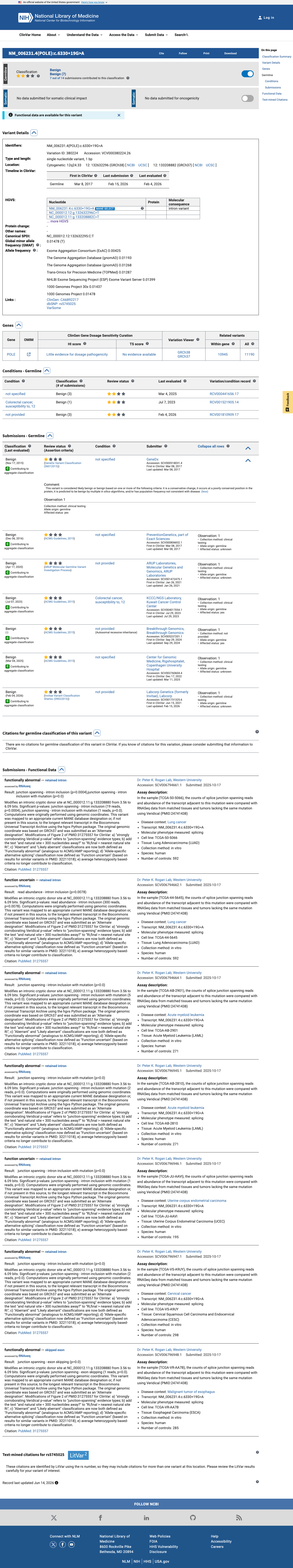
ClinVar
This variant has been reported in ClinVar as Benign (6 clinical laboratories). (ClinVarID = 380224)
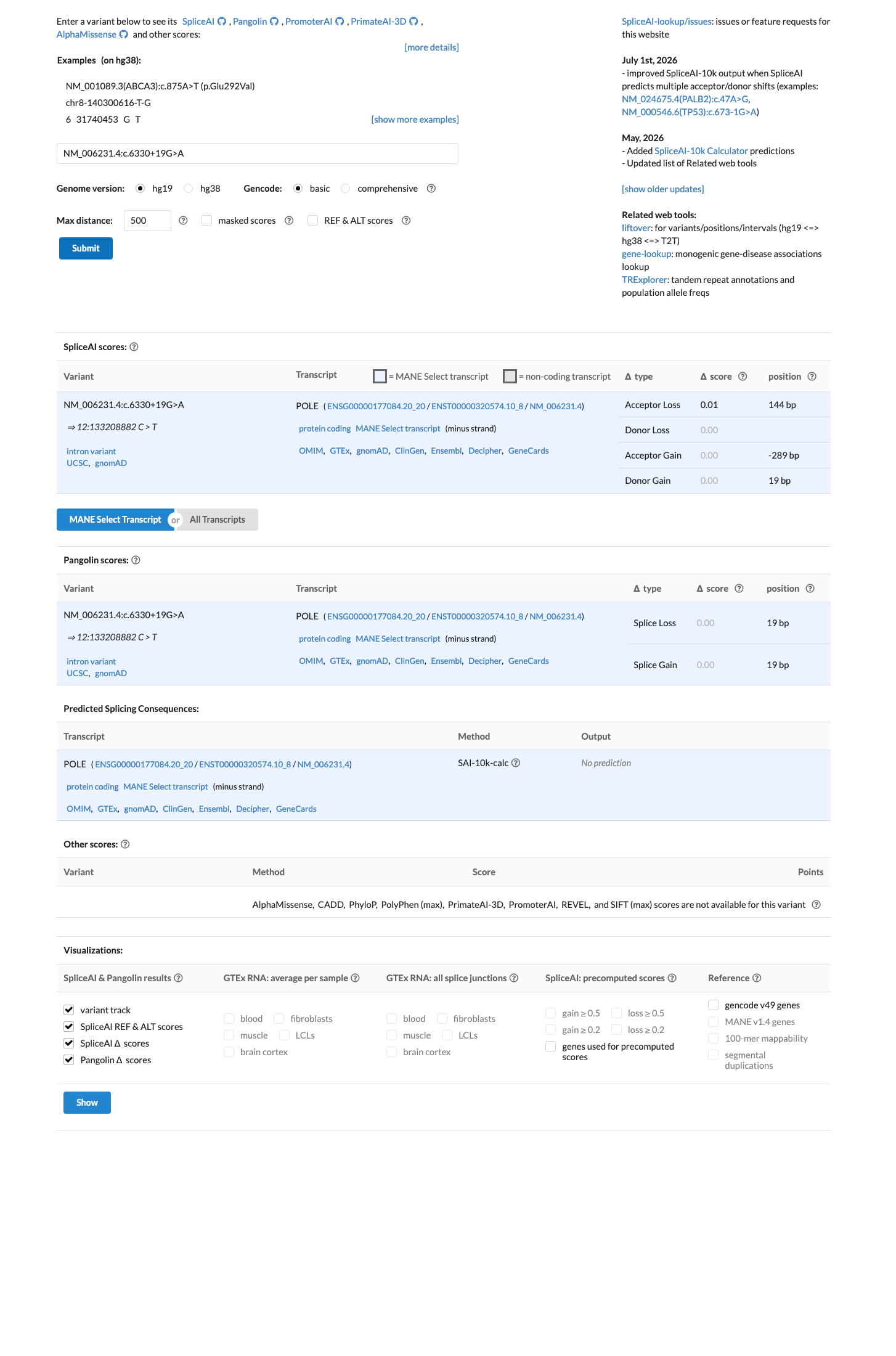
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR