PVS1_Strong: NM_001127208.2:c.5481del is a frameshift deletion (p.Lys1827AsnfsTer6) in TET2, a gene where germline loss of function is an established disease mechanism for ALPS-like immunodeficiency and hematologic malignancy predisposition. The variant resides in the terminal exon (exon 11/11) and is predicted to escape nonsense-mediated decay; strength is downgraded from PVS1 to PVS1_Strong per PMC6185798.1 PM2: Variant is entirely absent from gnomAD v4.1 (0/1,551,736 alleles, AF=0.0000%) and gnomAD v2.1, meeting the PM2 threshold for extremely low population frequency.2 No additional pathogenic or benign criteria were met. PS3 (functional evidence) and BS3 (benign functional evidence) could not be assessed as no variant-specific functional studies for NM_001127208.2:c.5481del were identified in the reviewed literature (PMID:21057493, PMID:24315485).3
TET2
Final classification
Likely Pathogenic
TET2 c.5481del · p.Lys1827AsnfsTer6
TET2
PVS1_Strong: NM_001127208.2:c.5481del is a frameshift deletion (p.Lys1827AsnfsTer6) in TET2, a gene where germline loss of function is an established disease mechanism for ALPS-like immunodeficiency and hematologic malignancy predisposition. The variant resides in the terminal exon (exon 11/11) and is predicted to escape nonsense-mediated decay; strength is downgraded from PVS1 to PVS1_Strong per PMC6185798.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PM2 moderate; combination = 1 strong + 1 moderate, which maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
TET2 c.5481del
PVS1 + PM2
→
Likely Pathogenic
1
pvs1_generic_framework ↗pvs1_gene_contextpvs1_variant_assessment
3
oncokb ↗
Gene diagram
· NM_001127208.2 · variants mapped to exon structure
TET2
NM_001127208.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 18 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
review
Pathogenic
NM_001127208.2:c.5481del is a frameshift deletion in exon 11 (the terminal exon) of TET2, predicted to cause p.Lys1827AsnfsTer6, a premature termination codon truncating the protein from 2003 to 1832 amino acids with loss of approximately 176 C-terminal residues. TET2 loss of function is a well-established germline disease mechanism (ALPS-like phenotype, hematologic malignancy predisposition). Under PMC6185798 (ClinGen SVI PVS1 recommendations), a null variant in a gene with established LOF disease mechanism qualifies for PVS1 at very strong level; however, because the variant resides in the terminal exon and is predicted to escape nonsense-mediated decay, the strength is downgraded to PVS1_Strong.
Frameshift deletion c.5481del predicted to cause p.Lys1827AsnfsTer6Variant in terminal exon (exon 11/11)NMD predicted to escape (PMC6185798 downgrade)
✓
PM2
moderate
Pathogenic
NM_001127208.2:c.5481del is entirely absent from gnomAD population databases: absent from v2.1 exomes, v4.1 exomes (0/1,399,432 alleles) and genomes (0/152,304 alleles), combined 0/1,551,736 alleles (AF=0.0000%). This meets the PM2 threshold for extremely low or absent population frequency (<0.1%).
gnomAD v4.1: 0/1551736 total alleles (AF=0.0000%)
Assessed · not applied
Pathogenic
PS2
No de novo evidence for this variant was identified.
PS3
No variant-specific functional studies for NM_001127208.2:c.5481del (p.Lys1827AsnfsTer6) were identified.
PS4
The variant is entirely absent from gnomAD v2.1, gnomAD v4.1 (0/1,551,736 alleles), and gnomAD-Canada.
PM1
Residue 1827 does not lie in a statistically significant cancer hotspot.
PM6
No de novo evidence was identified.
PP1
No segregation data are available for this variant.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No patient phenotype or family history data are available for this variant.
PP5
No reputable source has classified this variant as pathogenic.
Benign
BA1
The variant is entirely absent from gnomAD (0/1,551,736 alleles, AF=0.0000%), far below the BA1 threshold of >1% population frequency.
BS1
The variant is entirely absent from gnomAD (0/1,551,736 alleles, AF=0.0000%), far below the BS1 threshold of >0.3% population frequency.
BS2
No homozygous observations have been reported in any population database.
BS3
No variant-specific functional studies demonstrating a benign effect were identified.
BS4
No evidence of lack of segregation is available.
BP2
No observation of this variant in trans with a known pathogenic variant has been reported.
BP4
Multiple lines of computational evidence do not support a benign impact.
BP5
No alternative molecular basis for disease has been identified in this case.
BP6
No reputable source has classified this variant as benign.
N/A · 7
PS1 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
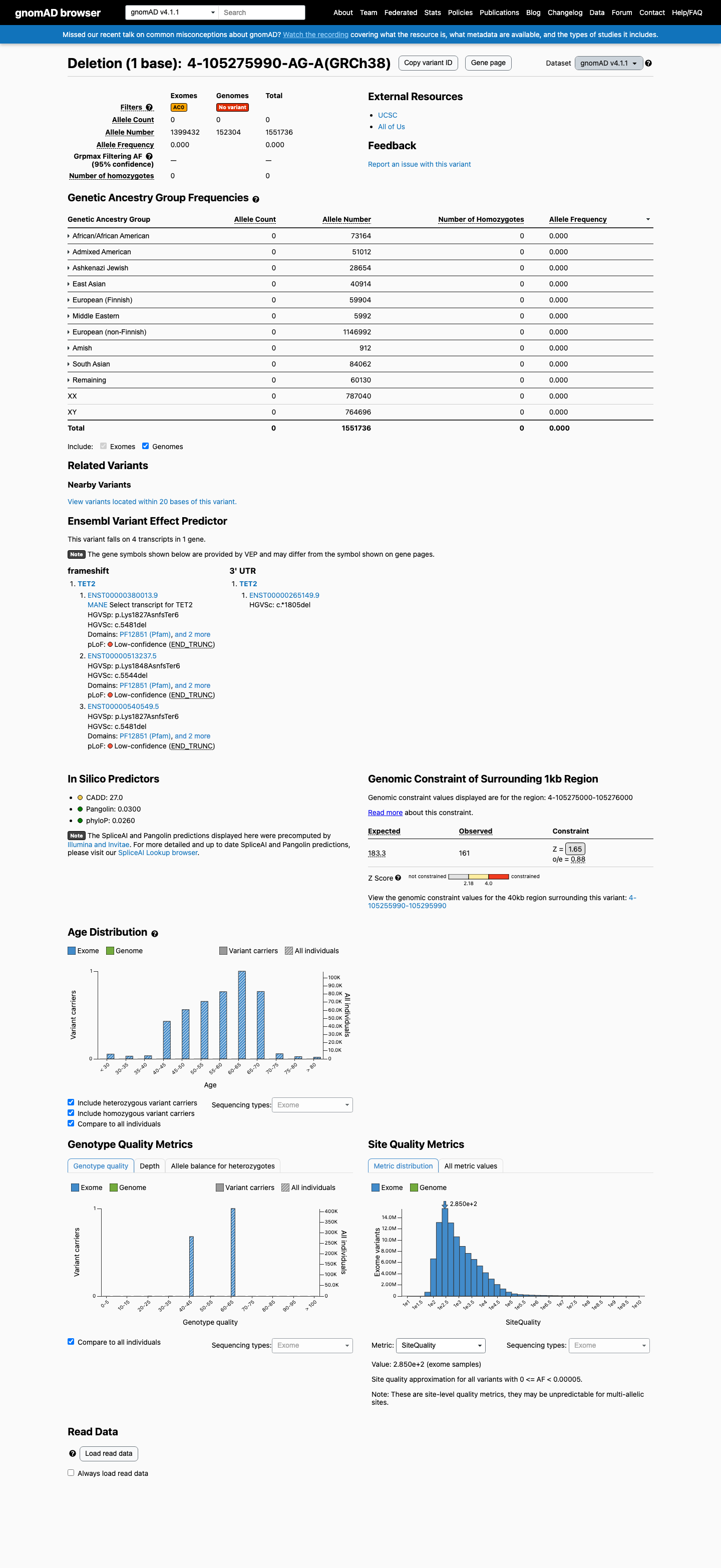
This variant is present in gnomAD v4.1 (AF= 0; MAF= 0.00000%, 0/1551736 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/73164 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / 1,551,736
0 hom
0 hom
Not observed in any ancestry group.
+ 10 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
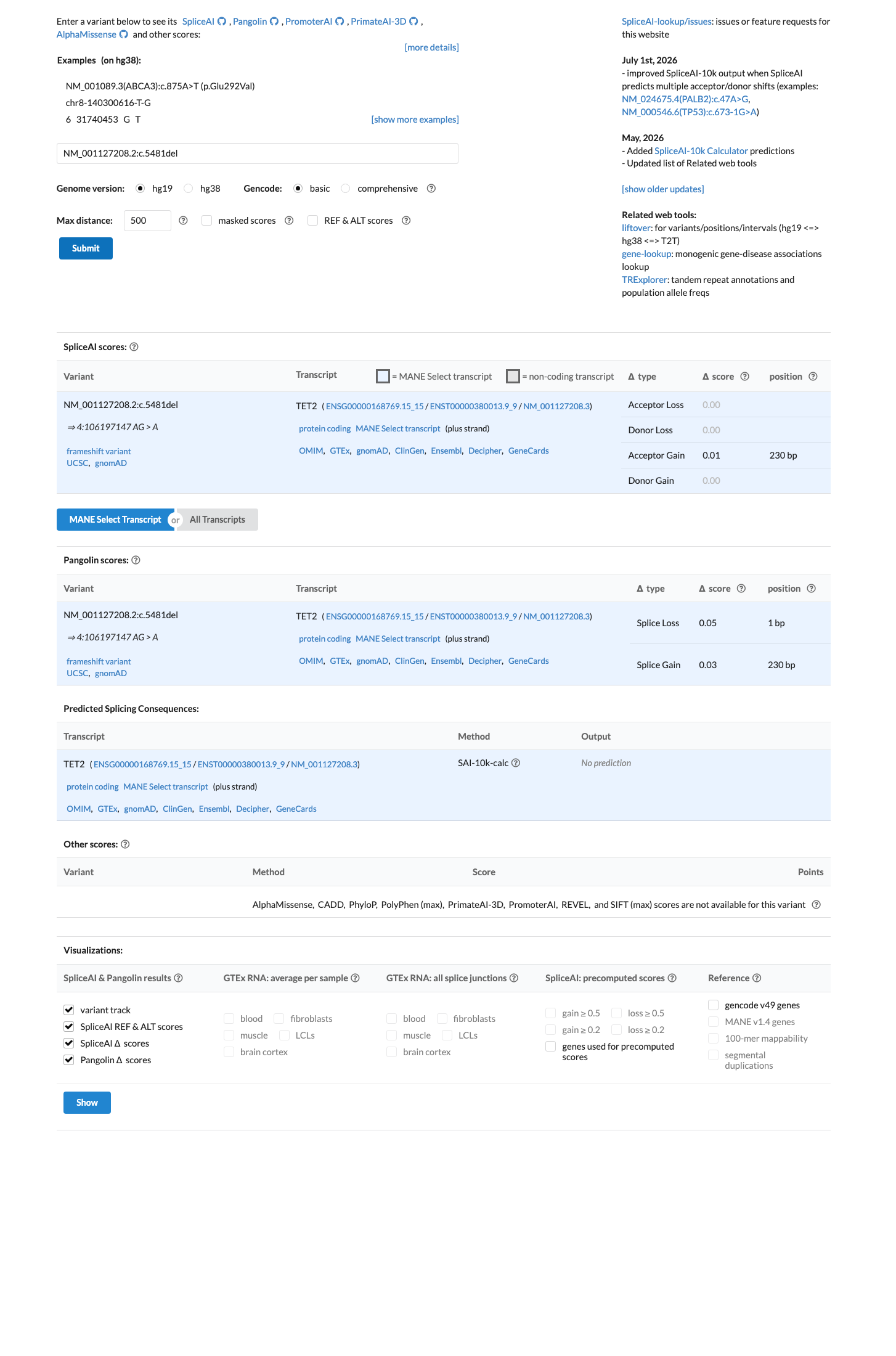
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
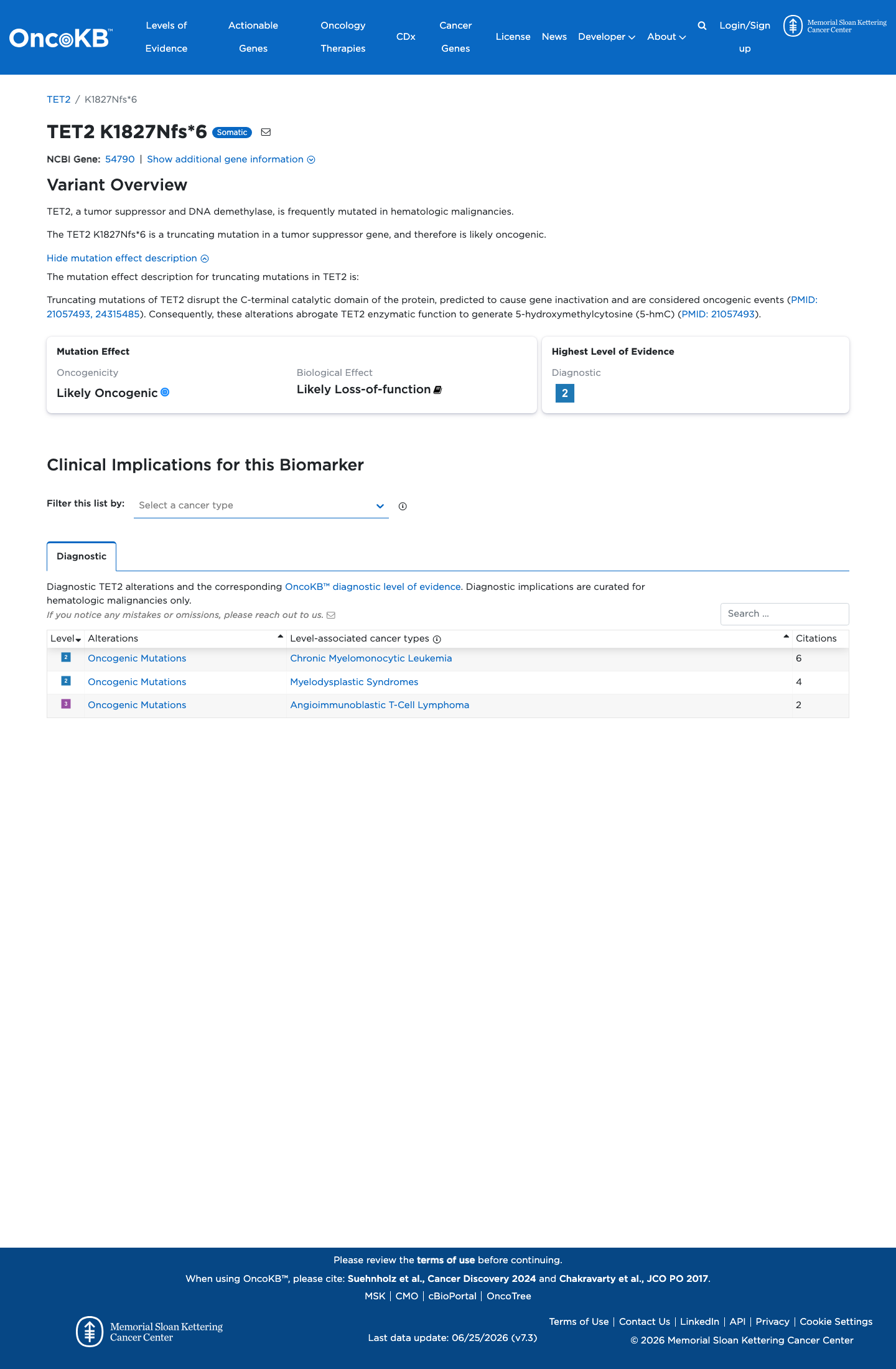
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
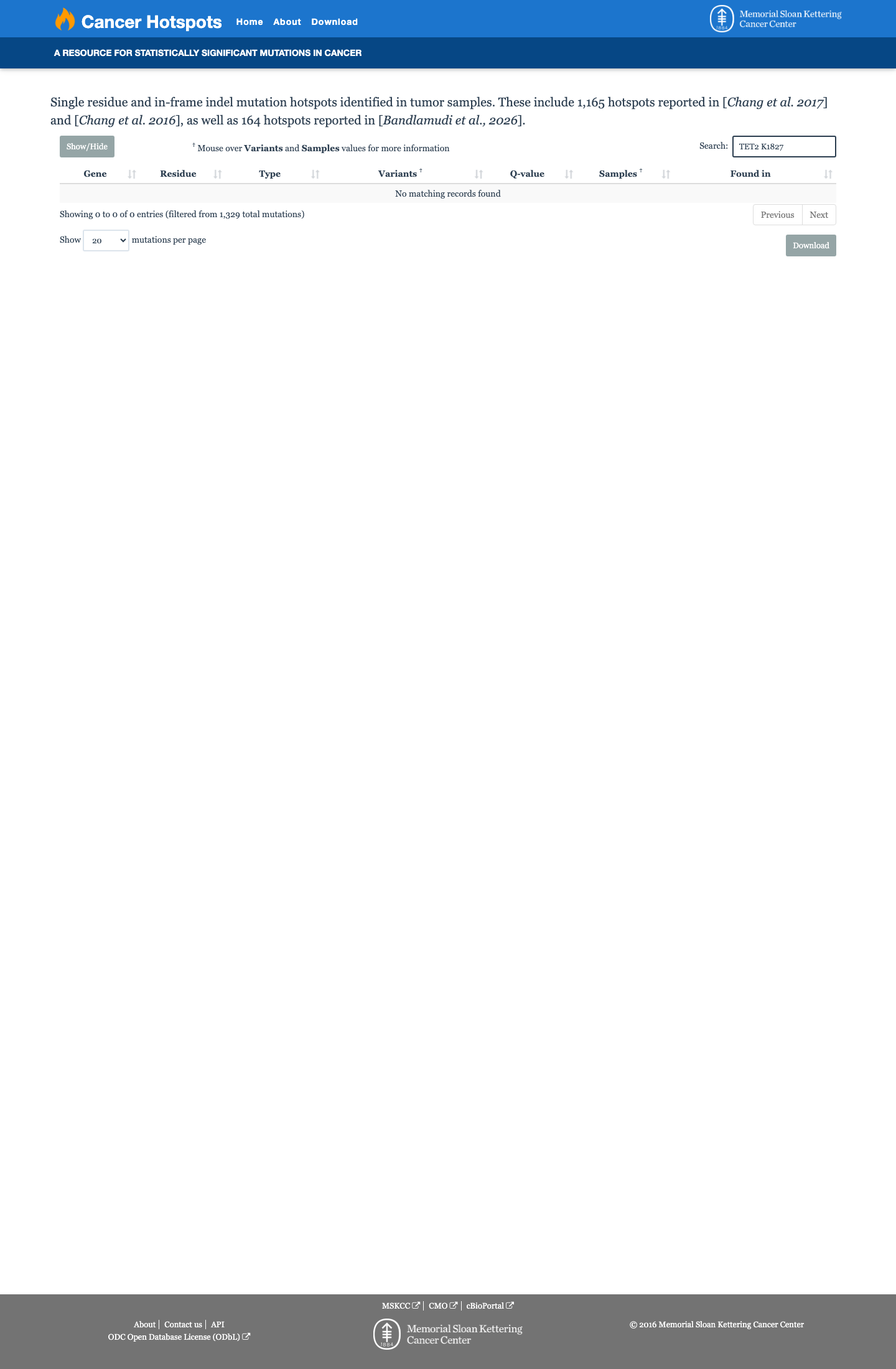
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 2 PMIDs not cited in assessment