NM_005475.2:c.127C>T (p.Arg43Cys) is a missense variant in SH2B3, a gene for which loss of function is a supported germline disease mechanism in myeloid and myeloproliferative disorders.1 This variant is present in gnomAD v4.1 at a total allele frequency of 0.045% (695/1,540,252 alleles) and in v2.1 at 0.024% (41/172,716 alleles), with no homozygotes observed. The highest subpopulation frequency is 0.059% in the European (non-Finnish) population. These frequencies fall below the 0.1% threshold for PM2 at supporting level.2 Multiple in silico tools predict a benign effect: REVEL score 0.254 (below pathogenic threshold), BayesDel score -0.346 (predicts benign), and SpliceAI max delta 0.00 (no splicing impact). This meets BP4 at supporting benign level.3 This variant is classified as Uncertain significance in ClinVar (1 submitter, GeneDx). OncoKB reports an Unknown Oncogenic Effect with no variant-specific curated functional evidence. No publications specifically mentioning NM_005475.2:c.127C>T were identified.4 With one supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4), the evidence is balanced toward neither pathogenic nor benign. Under generic ACMG/AMP 2015 combination rules, this does not meet the threshold for Likely Pathogenic, Likely Benign, Pathogenic, or Benign. The variant remains a Variant of Uncertain Significance.5
SH2B3
Final classification
VUS
SH2B3 c.127C>T · p.Arg43Cys
SH2B3
NM_005475.2:c.127C>T (p.Arg43Cys) is a missense variant in SH2B3, a gene for which loss of function is a supported germline disease mechanism in myeloid and myeloproliferative disorders.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
SH2B3 c.127C>T
PM2 + BP4
→
VUS
Gene diagram
· NM_005475.2 · variants mapped to exon structure
SH2B3
NM_005475.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 10 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
PM2 (supporting) is met: NM_005475.2:c.127C>T is present in gnomAD at a total allele frequency of 0.045% (695/1,540,252 alleles in v4.1; 41/172,716 in v2.1), which falls below the 0.1% threshold for PM2. Homozygotes are absent. However, the presence of 695 alleles across gnomAD v4.1 indicates this is not an ultra-rare variant, limiting strength to supporting.
gnomAD v4.1: AF 0.045% (695/1540252 alleles
✓
BP4
supporting
Benign
BP4 (supporting benign) is met: multiple lines of computational evidence suggest NM_005475.2:c.127C>T (p.Arg43Cys) has no deleterious impact. REVEL score is 0.254 (below pathogenic threshold), BayesDel score is -0.346 (predicts benign), and SpliceAI predicts no splicing alteration (max delta 0.00). All three independent in silico tools concur in predicting a neutral effect.
REVEL score: 0.254 (below 0.5 pathogenic thresholdpredicts benign)BayesDel score: -0.346 (negative score
Assessed · not applied
Pathogenic
PS2
PS2 cannot be assessed: no de novo data are available.
PS3
PS3 cannot be assessed: no variant-specific functional studies were identified for NM_005475.2:c.127C>T (p.Arg43Cys).
PM6
PM6 cannot be assessed: no de novo data are available for NM_005475.2:c.127C>T.
PP1
PP1 cannot be assessed: no segregation data are available for NM_005475.2:c.127C>T.
PP3
PP3 is not met: multiple in silico tools predict a benign effect for NM_005475.2:c.127C>T (p.Arg43Cys).
Benign
BA1
BA1 is not met: the highest observed population allele frequency is 0.059% (European non-Finnish in gnomAD v4.1), which is far below the 1% BA1 threshold.
BS1
BS1 is not met: the highest observed population allele frequency is 0.059% (European non-Finnish in gnomAD v4.1), which is below the 0.3% BS1 threshold.
BS2
BS2 cannot be assessed: although NM_005475.2:c.127C>T is observed in 695 heterozygous individuals in gnomAD v4.1, no phenotype data are available for these carriers.
BS3
BS3 cannot be assessed: no variant-specific functional studies demonstrating a neutral or benign effect for NM_005475.2:c.127C>T are available.
BS4
BS4 cannot be assessed: no segregation data demonstrating lack of co-segregation with disease are available for NM_005475.2:c.127C>T.
N/A · 13
PVS1 · PS1 · PS4 · PM1 · PM5 · PP2 · PP4 · PP5 · BP1 · BP2 · BP5 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
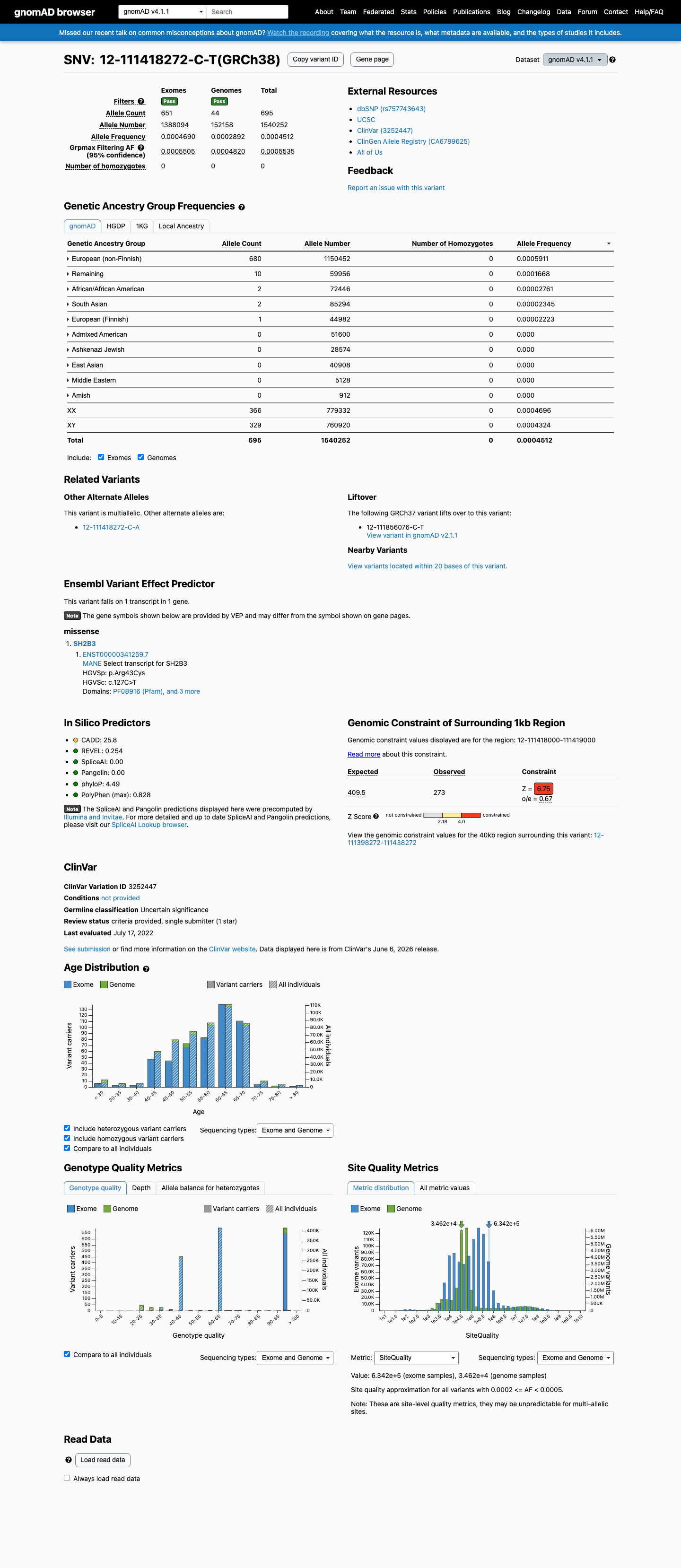
This variant is present in gnomAD v4.1 (AF= 0.000451225; MAF= 0.04512%, 695/1540252 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000591072; MAF= 0.05911%, 680/1150452 alleles, homozygotes = 0); grpmax FAF= 0.00055351.
v2.1
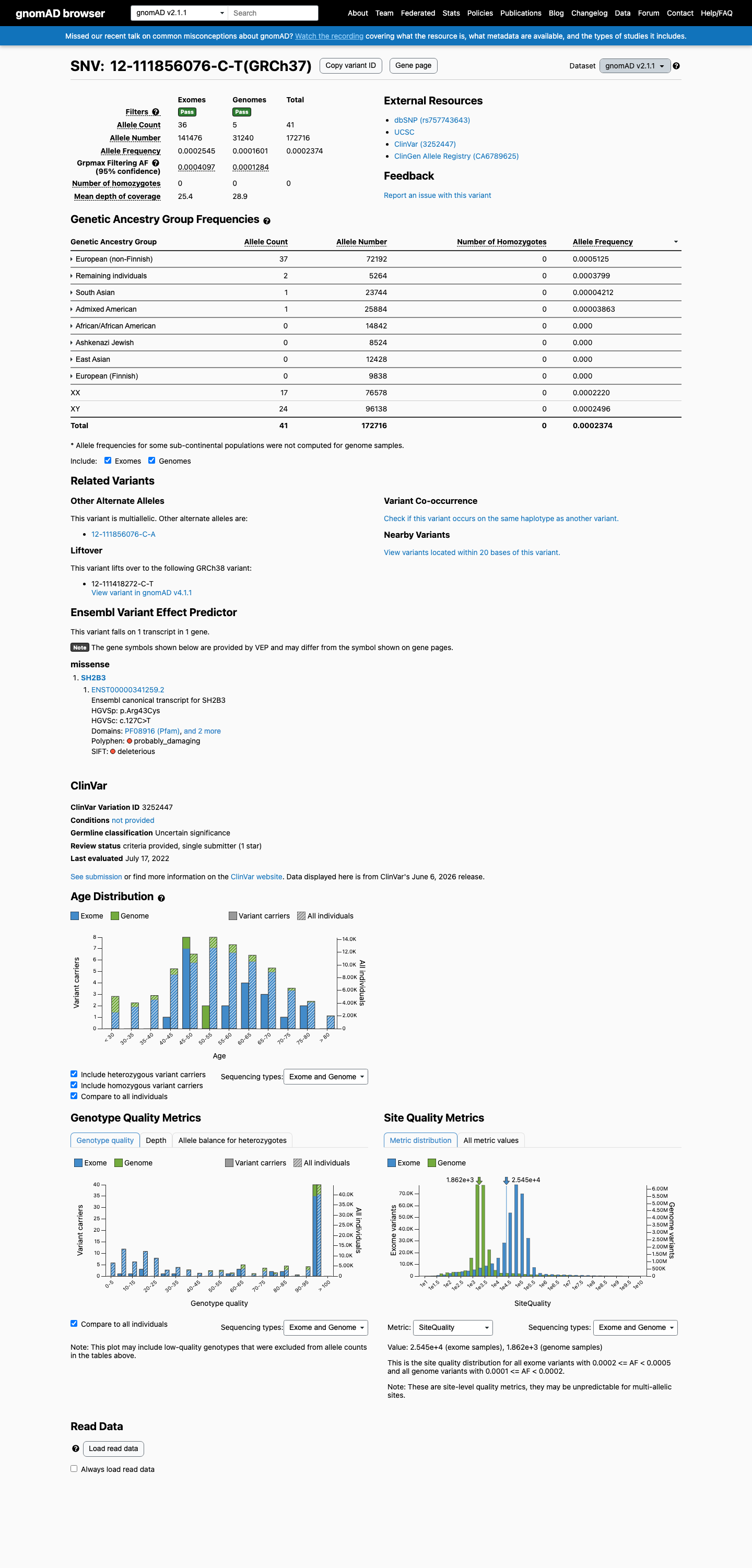
This variant is present in gnomAD v2.1 (AF= 0.000237384; MAF= 0.02374%, 41/172716 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000512522; MAF= 0.05125%, 37/72192 alleles, homozygotes = 0); grpmax FAF= 0.00040972.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00016286644951140066, 3/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.045%
· 695 / 1,540,252
0 hom · FAF 0.055%
0 hom · FAF 0.055%
European (non-Finnish) 680 / 1,150,452 |
0.059% |
Remaining individuals 10 / 59,956 |
0.017% |
African/African American 2 / 72,446 |
0.0028% |
South Asian 2 / 85,294 |
0.0023% |
European (Finnish) 1 / 44,982 |
0.0022% |
+ 5 not observed (Admixed American, Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.024%
· 41 / 172,716
0 hom · FAF 0.041%
0 hom · FAF 0.041%
European (non-Finnish) 37 / 72,192 |
0.051% |
Remaining individuals 2 / 5,264 |
0.038% |
South Asian 1 / 23,744 |
0.0042% |
Admixed American 1 / 25,884 |
0.0039% |
+ 4 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
0.016%
· 3 / 18,420
0 hom · FAF 0.0069%
0 hom · FAF 0.0069%
indel · split
European (non-Finnish) 3 / 11,740 |
0.026% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
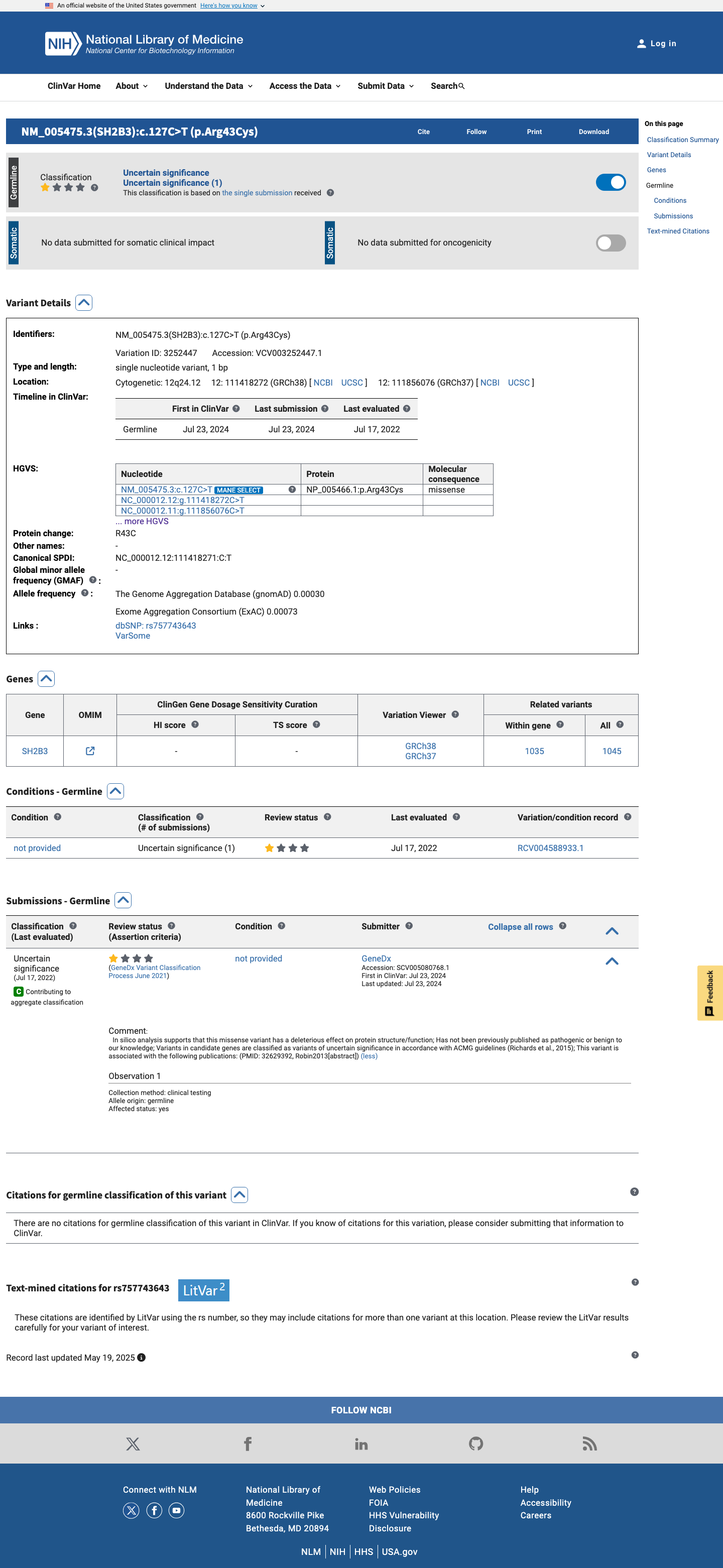
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory). (ClinVarID = 3252447)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.254. BayesDel score = -0.345922.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. SH2B3 is an adaptor protein that regulates growth factor and cytokine signaling. Mutations are found in hematopoietic disorders including leukemias an
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
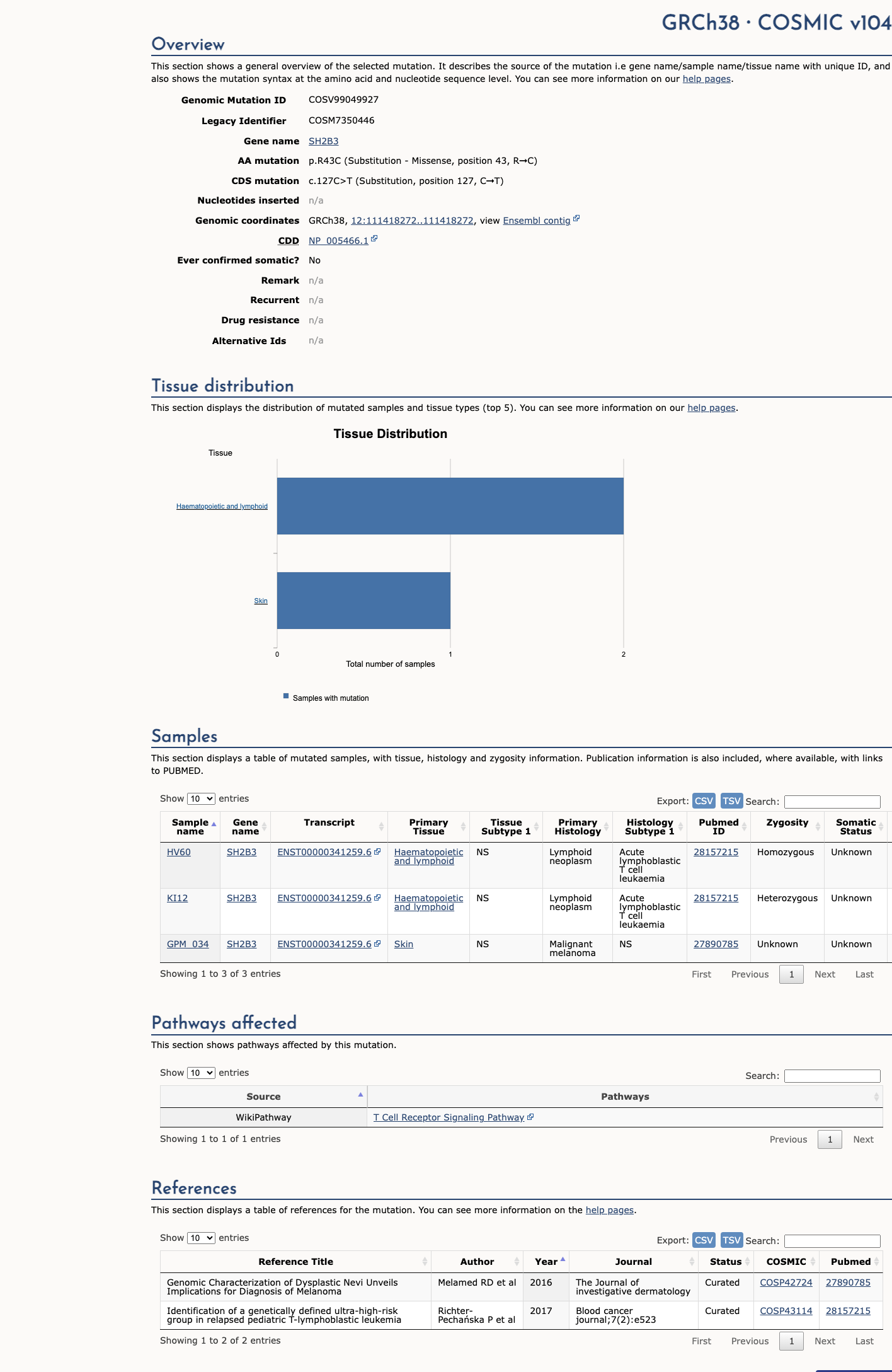
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99049927, n = 3 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links