Classification rationale
PVS1PM2
Likely Pathogenic
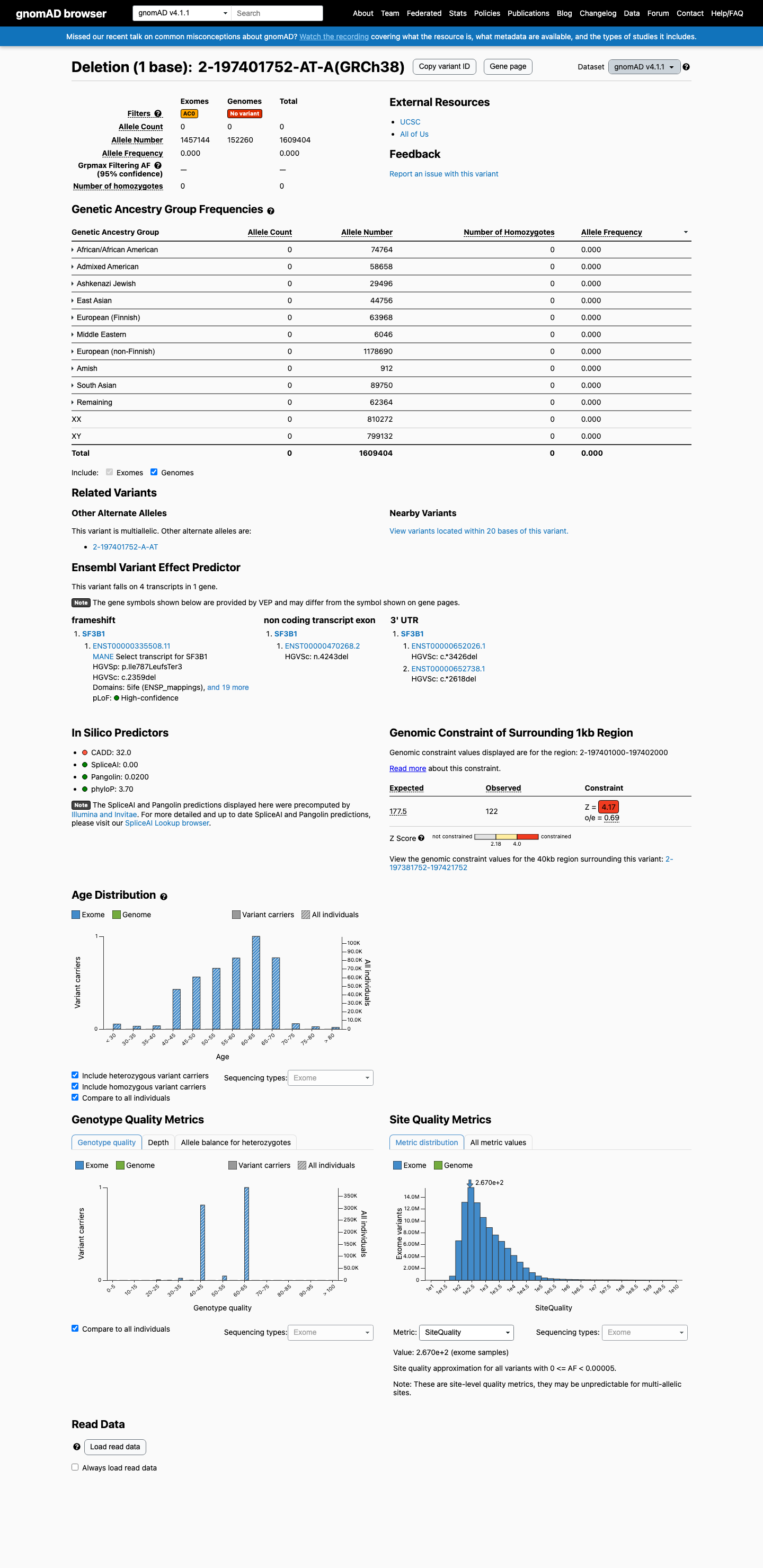
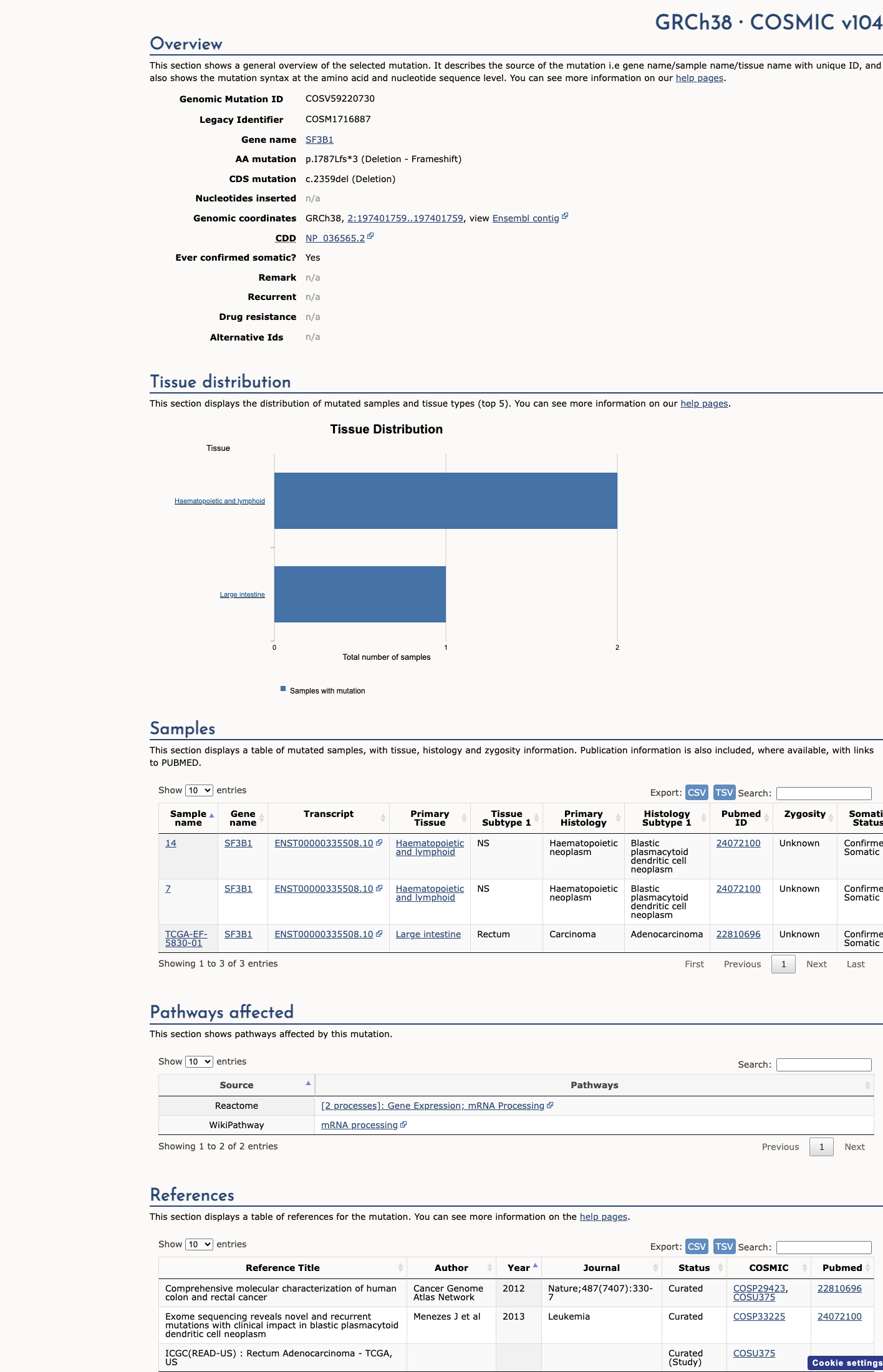
SF3B1 c.2359del
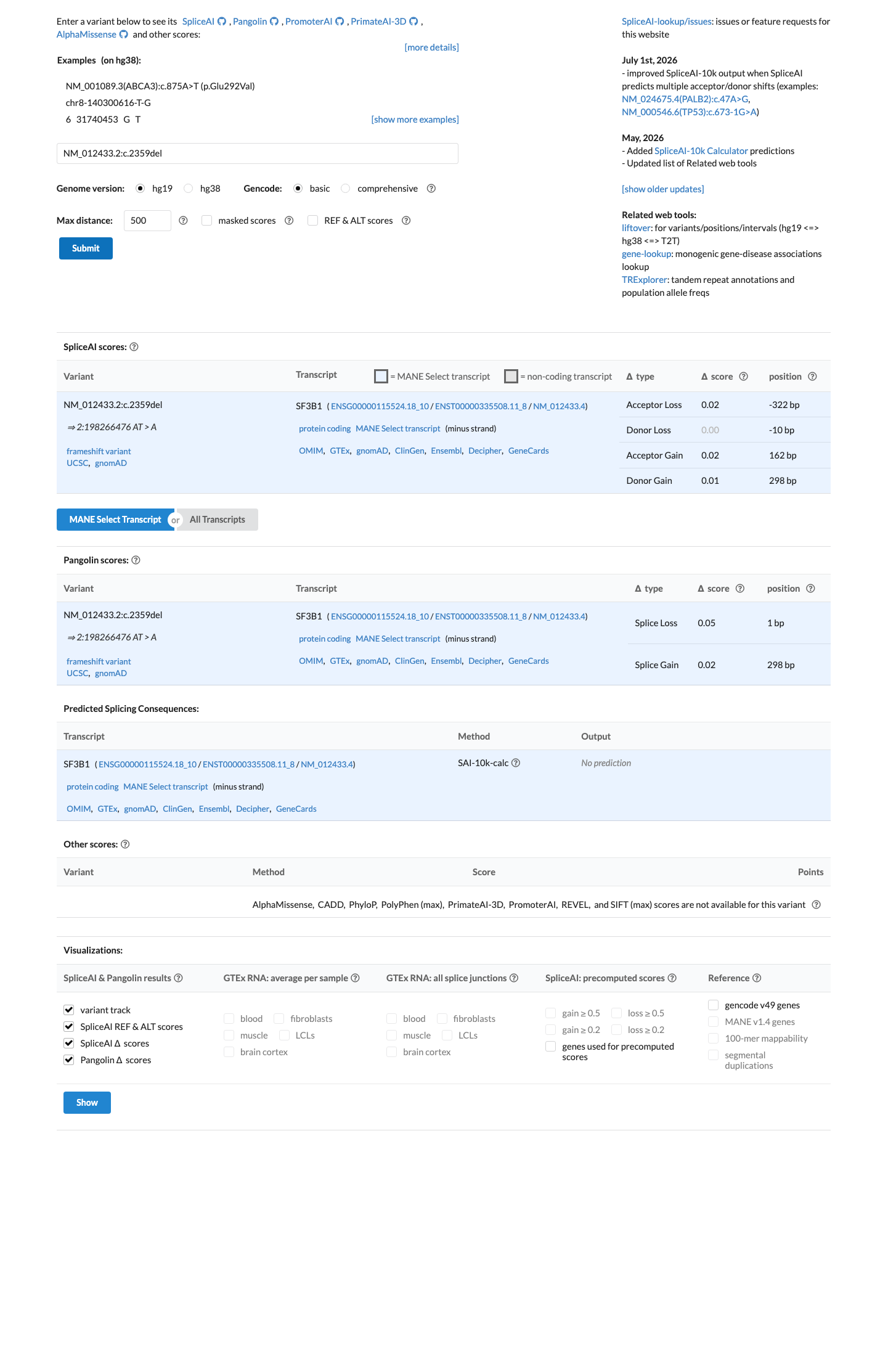
PVS1 (very_strong): NM_012433.2:c.2359del is a frameshift deletion predicted to cause premature termination (p.Ile787LeufsTer3) in SF3B1, a gene in which germline loss of function is an established disease mechanism for neurodevelopmental disorders (PMC6185798; PMID:41577671).1 PM2 (moderate): This variant is absent from large population databases including gnomAD v2.1 and v4.1 (0/1,609,404 alleles), consistent with rarity in the general population.2 Classification of Likely Pathogenic is assigned based on 1 very strong criterion (PVS1) and 1 moderate criterion (PM2) under the generic ACMG/AMP 2015 combination rules (PMID:25741868).3
PVS1 + PM2
→
Likely Pathogenic
1
pvs1_generic_framework ↗pvs1_variant_assessment
3
generic_acmg_combination_rules