NM_004656.4:c.86T>G (p.Val29Gly) in BAP1 is a missense variant in exon 3, located within the ubiquitin C-terminal hydrolase (UCH) domain.1 This variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada, meeting PM2 at moderate strength.2 The variant resides in the UCH domain, a critical functional domain of BAP1 without evidence of benign variation at this residue, meeting PM1 at moderate strength as assigned by the Walpole et al. 2018 global cohort study.3 Computational evidence supports a deleterious effect: REVEL score 0.77 exceeds commonly used thresholds, and Walpole et al. explicitly assigned PP3 to p.V29G, meeting PP3 at supporting strength.4 A saturation genome editing study of BAP1 (PMID:38969833) was reviewed in full but did not include p.Val29Gly; the Ambry Genetics ClinVar PS3 functional signal citing this paper could not be validated.5 No variant-specific functional studies, de novo observations, or segregation data sufficient to meet PS3, PS2, or PP1 were identified. With two moderate criteria (PM1, PM2) and one supporting criterion (PP3) met, and no benign criteria met, this variant does not reach the likely pathogenic threshold (requires ≥3 moderate, or 2 moderate + 2 supporting) under the generic ACMG/AMP 2015 classification framework and is classified as a Variant of Uncertain Significance (VUS).6
BAP1
Final classification
VUS
BAP1 c.86T>G · p.Val29Gly
BAP1
NM_004656.4:c.86T>G (p.Val29Gly) in BAP1 is a missense variant in exon 3, located within the ubiquitin C-terminal hydrolase (UCH) domain.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 moderate, PM2 moderate, PP3 supporting; combination = 2 moderate + 1 supporting, which maps to VUS.
Classification rationale
PM1PM2PP3
VUS
BAP1 c.86T>G
PM1 + PM2 + PP3
→
VUS
4
revelPMID:30517737 ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_004656.4 · variants mapped to exon structure
BAP1
NM_004656.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 19 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
p.Val29Gly is located in the ubiquitin C-terminal hydrolase (UCH) domain of BAP1, a well-established critical functional domain. The variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada, indicating no benign variation at this residue. PMID:30517737 explicitly assigns PM1 to p.V29G in its ACMG assessment.
PMID:30517737 line 255-259: 'all have PM1 and PP3.' UCH domain (amino acids 1-240) houses all nine missense variants with evidence of pathogenicity. Absent from all population databases.
✓
PM2
moderate
Pathogenic
Absent from gnomAD v2.1 (exomes), gnomAD v4.1 (exomes+genomes), and gnomAD-Canada v1.0 (genomes). Allele frequency is below the PM2 threshold of <0.1% in all population databases examined.
Absent from gnomAD v2.1gnomAD v4.1and gnomAD-Canada v1.0. AF = 0.0
✓
PP3
supporting
Pathogenic
Multiple lines of computational evidence support a deleterious effect on the gene product. REVEL score is 0.77, exceeding the 0.5 and 0.75 thresholds used by many clinical laboratories. PMID:30517737 explicitly assigns PP3 to p.V29G. SpliceAI predicts no splice impact (max delta 0.03), which is not contradictory — this is a missense, not splice variant.
REVEL score 0.77 (deleterious). SpliceAI max delta 0.03 (no splice impactnot contradictory for missense). PMID:30517737 line 256: 'all have PM1 and PP3.' BayesDel 0.315 is below typical damaging threshold but does not outweigh the REVEL score and expert assignment.
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change at the same codon producing the same missense change (p.Val29Gly) with established pathogenicity.
PS2
PS2 requires de novo confirmation with both maternity and paternity confirmed.
PS3
Well-established functional studies supporting a damaging effect were not identified.
PS4
PS4 requires a statistically enriched observation of the variant in affected individuals versus controls.
PM6
PM6 requires a de novo observation with maternity and paternity confirmed.
PP1
PP1 requires co-segregation with disease in multiple affected family members.
PP2
PP2 applies to genes with a low rate of benign missense variation where missense variants are a common disease mechanism.
PP4
PP4 requires the variant to be observed in a patient whose phenotype or family history is highly specific for the disease.
PP5
No reputable source has definitively classified this variant as pathogenic.
Benign
BA1
BA1 requires allele frequency >1% in population databases.
BS1
BS1 requires allele frequency >0.3% in population databases.
BS2
BS2 requires observation in a healthy adult individual for a disorder with full penetrance expected at an early age.
BS3
BS3 requires well-established functional studies showing no damaging effect.
BS4
BS4 requires lack of segregation in affected family members.
BP1
BP1 applies when a missense variant occurs in a gene for which primarily truncating variants are known to cause disease.
BP2
BP2 requires observation in trans with a pathogenic variant for a fully penetrant dominant disorder, or in cis with a pathogenic variant.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on gene or gene product.
BP5
BP5 requires the variant to be found in a case with an alternate molecular basis for disease.
BP6
BP6 requires a reputable source to report the variant as benign.
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
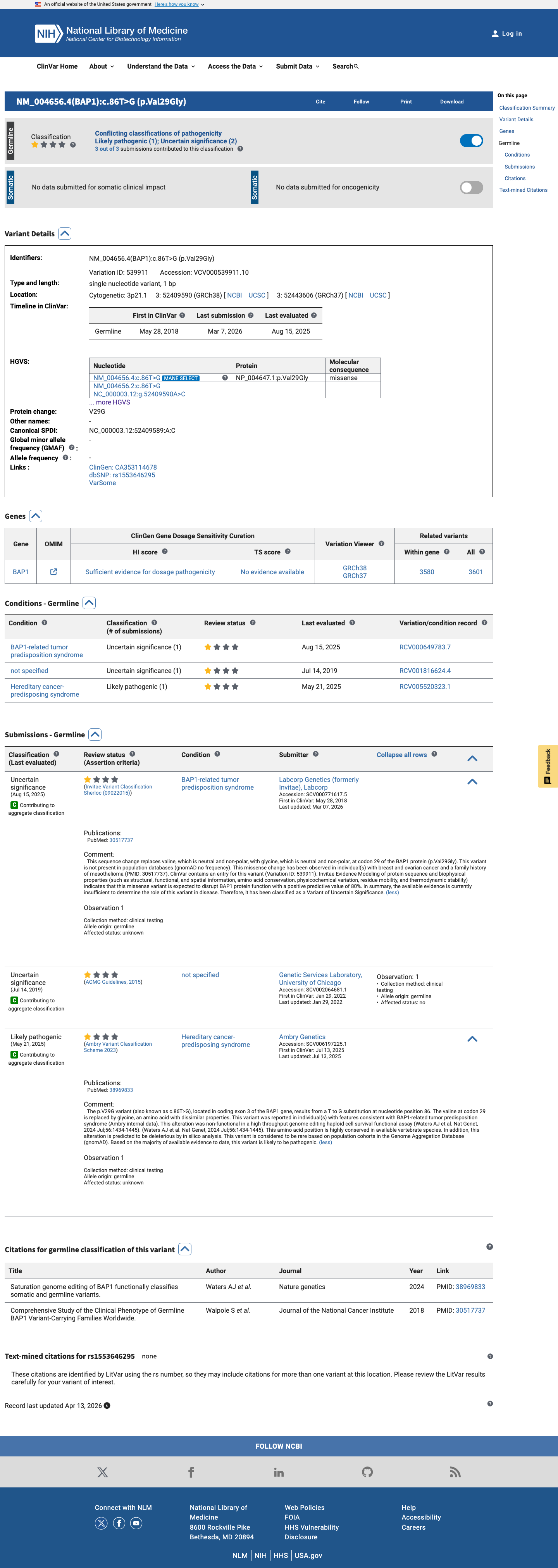
ClinVar
This variant has been reported in ClinVar as Uncertain significance (2 clinical laboratories) and as Likely pathogenic (1 clinical laboratory). (ClinVarID = 539911)
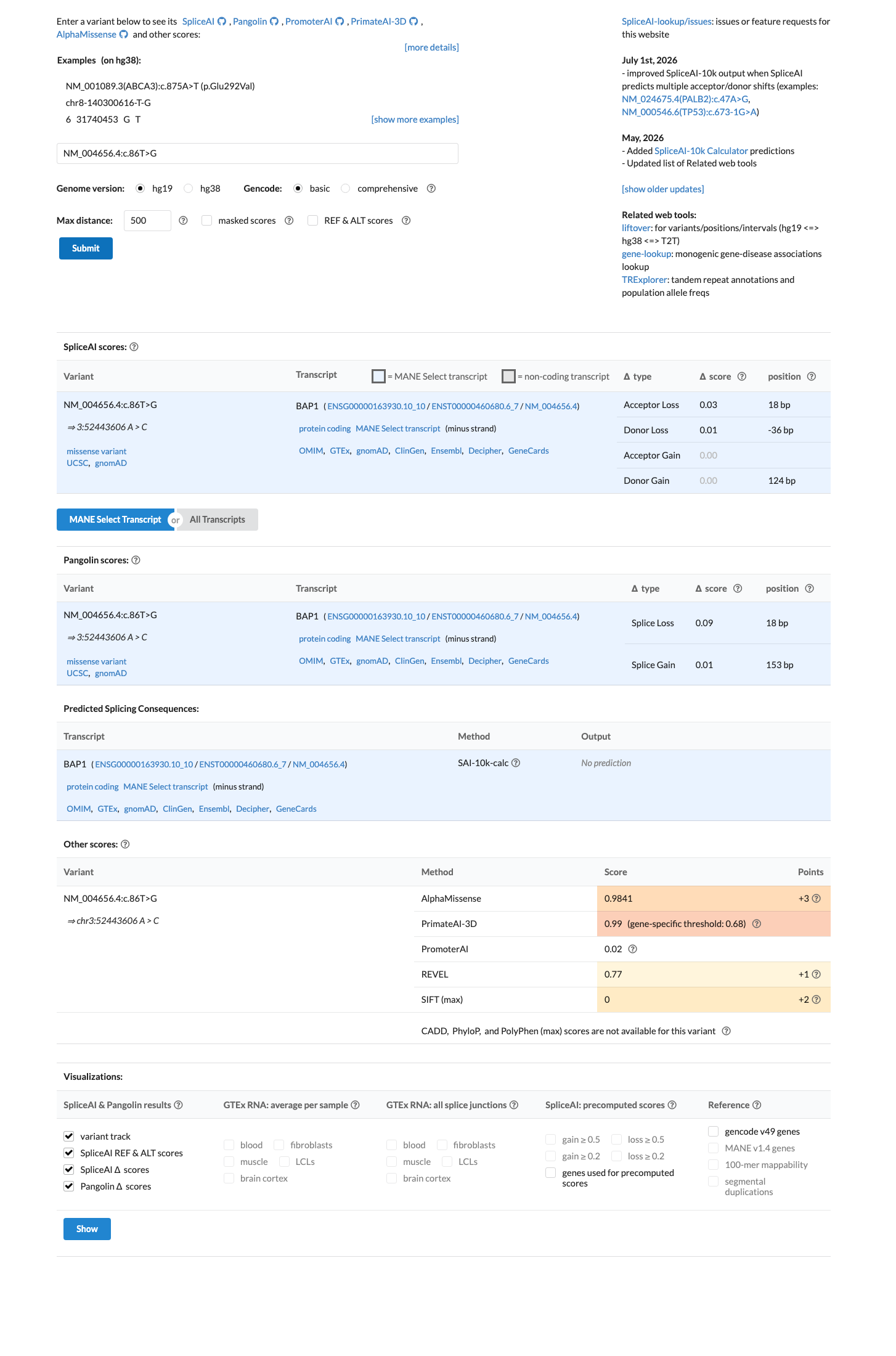
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03). REVEL score = 0.77. BayesDel score = 0.315276.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. BAP1, a nuclear ubiquitin hydrolase, is frequently altered by mutation in various cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 4 further PMIDs triaged but not cited — see Sources & References.
Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide.
Searched
c.86T>Gp.Val29GlyV29Gp.V29G
Found
p.V29G is identified among six missense variants with some evidence of pathogenicity. All six had PM1 and PP3; some also had varying levels of PP1 and PP4, though none reached the likely pathogenic ACMG/AMP threshold. All nine missense variants identified in this study with evidence of pathogenicity occur in the UCH domain.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PP3 supports · met
Why
Variant-specific evidence confirms location in the UCH functional domain supporting PM1, and explicit author assignment of PP3 supports computational evidence of a deleterious effect.
We also identified an additional six variants (p.L14H, p.V29G, p.D67G, p.N78S, p.L180P, p.W202R) that have some evidence of pathogenicity (ie, all have PM1 and PP3 and some have varying levels of PP1 and PP4) but do not reach the likely pathogenic ACMG threshold
Location Results, paragraphs on missense variant classification (lines 248-259) and Discussion (lines 808-818) · full text
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
38969833 ↗
Saturation genome editing of BAP1 functionally classifies somatic and germline variants.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR