NM_006231.4:c.1871A>C (p.His624Pro) is a missense variant in exon 19 of POLE, located in the C-terminal polymerase domain outside the exonuclease domain (residues ~268–471) where established pathogenic hotspot variants cluster.1 This variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada, comprising over 300,000 population alleles without observation (PM2_Supporting).2 The variant has been reported in ClinVar as Uncertain significance by a single clinical laboratory (Labcorp Genetics, SCV001206773), and no additional submissions from expert panels are available.3 The León-Castillo et al. 2020 custom POLE framework does not assign PM1, PS4, PP3, or BP4 to this variant, as p.His624Pro is absent from all supplementary tables (S1, S2, S3) and is not among the established exonuclease-domain hotspot or recurrent variants.4 Computational analyses are equivocal: REVEL score 0.712 is borderline; BayesDel score 0.151 is modest; SpliceAI predicts no splicing impact (max delta 0.01). These do not meet thresholds for PP3 or BP4 under either the custom or generic frameworks.5 No variant-specific functional studies, segregation data, de novo observations, case-control enrichment, or phenotype specificity data were identified in the literature or public databases.6 With only PM2_Supporting met, this variant does not reach the threshold for Likely Pathogenic (requires ≥2 Supporting with at least PVS1, or 1 Moderate + 4 Supporting, etc.) or Likely Benign (requires ≥2 Supporting benign). The variant remains classified as a Variant of Uncertain Significance (VUS) under ACMG/AMP 2015 combination rules.7
POLE
Final classification
VUS
POLE c.1871A>C · p.His624Pro
POLE
NM_006231.4:c.1871A>C (p.His624Pro) is a missense variant in exon 19 of POLE, located in the C-terminal polymerase domain outside the exonuclease domain (residues ~268–471) where established pathogenic hotspot variants cluster.
Only one supporting-level pathogenic criterion (PM2_Supporting) is met. The León-Castillo 2020 custom POLE framework adopts generic ACMG/AMP 2015 final combination rules, under which a single supporting criterion is insufficient to reach Likely Pathogenic (minimum: PVS1 + 2 Supporting, 1 Strong + 2 Supporting, 3 Moderate, 2 Moderate + 2 Supporting, or 1 Moderate + 4 Supporting), nor does any benign-classification combination apply. The variant defaults to VUS.
Classification rationale
PM2
VUS
POLE c.1871A>C
PM2
→
VUS
1
vcep_path_250_323vcep_path_250_323_s002
4
vcep_path_250_323_s002vcep_path_250_323_s003vcep_path_250_323_s004
5
revelbayesdelspliceai ↗
6
oncokb ↗
7
generic_acmg_combination_rules
Gene diagram
· NM_006231.4 · variants mapped to exon structure
POLE
NM_006231.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 22 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_006231.4:c.1871A>C (p.His624Pro) is absent from gnomAD v2.1 (exomes), gnomAD v4.1 (exomes), and gnomAD-Canada v1.0 (genomes), comprising a total population of over 300,000 alleles without observation. This supports a pathogenic role for a rare missense variant in a gene where pathogenic variants are established.
Absent from gnomAD v2.1 (exomes).Absent from gnomAD v4.1 (exomes).Absent from gnomAD-Canada v1.0 (genomes).
Assessed · not applied
Pathogenic
PS1
No evidence of a different nucleotide change at codon 624 resulting in the same amino acid substitution (p.His624Pro) that has been previously classified as pathogenic.
PS2
No de novo occurrence data available for this variant.
PS3
No variant-specific functional evidence was identified.
PS4
The POLE custom framework (León-Castillo 2020) restricts PS4_Supporting to exact established pathogenic hotspot variants (P286R, V411L, A456P, S297F) that are recurrent in both COSMIC and TCGA endometrial carcinoma cohorts with combined EC count ≥10.
PM1
p.His624Pro is located at residue 624 in the C-terminal polymerase domain of POLE, outside the exonuclease domain (residues ~268–471) where established pathogenic hotspot variants cluster.
PM5
No same-residue (codon 624) comparator missense variant with a different amino acid change has been identified as pathogenic or likely pathogenic.
PM6
No de novo observation has been reported for this variant.
PP1
No segregation data are available for this variant.
PP2
No HCI prior probability score is available for POLE.
PP3
p.His624Pro is not present in the León-Castillo supplementary in silico tables (S2 or S3), so the custom PP3 rule does not apply.
PP4
No information is available regarding the clinical phenotype or specificity of presentation for this variant.
PP5
No reputable source has recently reported this variant as pathogenic.
Benign
BA1
NM_006231.4:c.1871A>C is absent from all gnomAD population databases (v2.1, v4.1, Canada).
BS1
NM_006231.4:c.1871A>C is absent from all gnomAD population databases.
BS2
No data are available regarding observation of this variant in healthy adult individuals in a context that would support a benign classification.
BS3
No functional evidence demonstrating a neutral or benign effect has been identified for this variant.
BS4
No segregation data are available for this variant.
BP1
BP1 applies when a missense variant occurs in a gene for which primarily truncating variants are known to cause disease.
BP2
No data are available regarding observation of this variant in trans with a known pathogenic variant in POLE.
BP4
p.His624Pro is not present in the León-Castillo supplementary in silico tables (S2 or S3), so the custom BP4 rule does not apply.
BP5
No evidence of an alternate molecular basis for disease has been identified in individuals carrying this variant.
BP6
No reputable source has reported this variant as benign or likely benign.
N/A · 5
PVS1 · PM3 · PM4 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
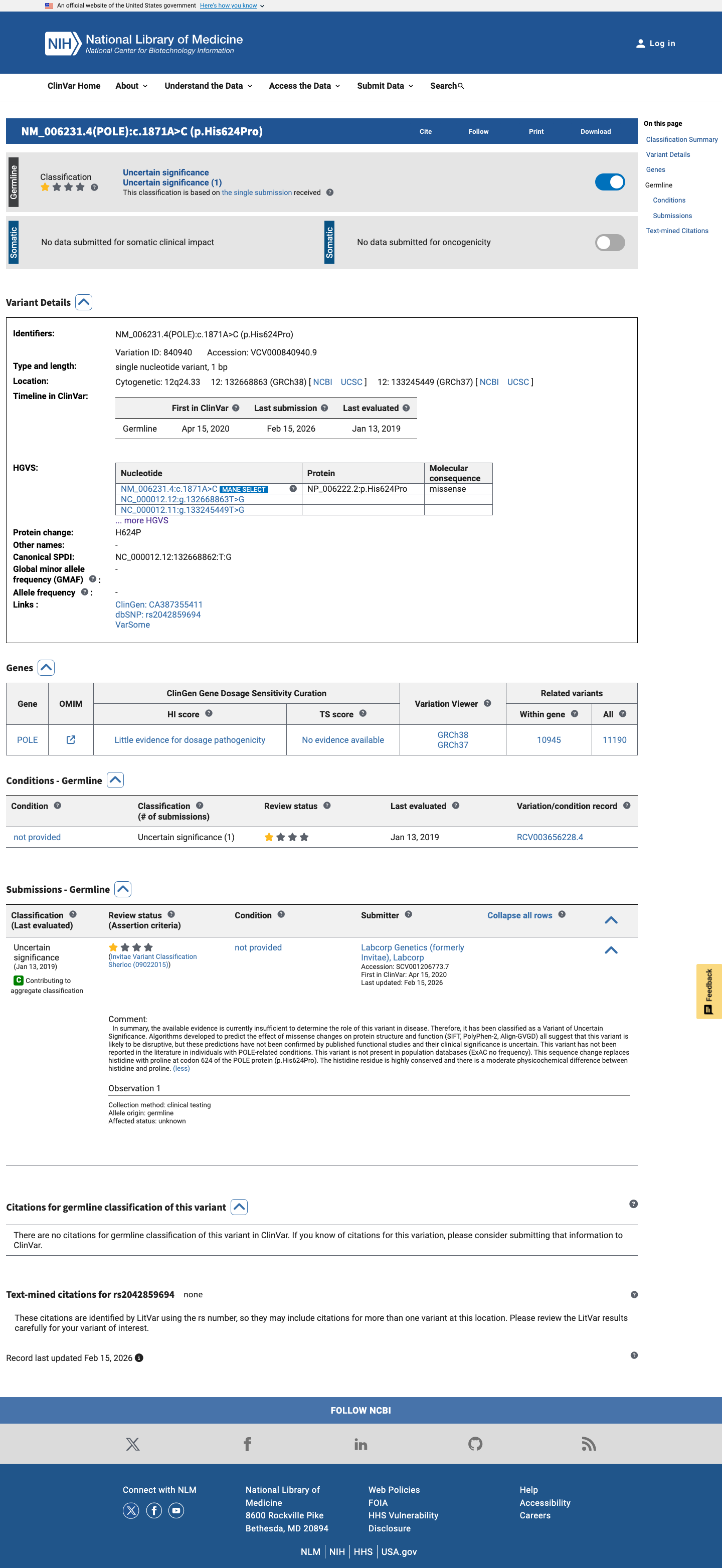
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory). (ClinVarID = 840940)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.712. BayesDel score = 0.150613.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. POLE, the catalytic subunit of DNA polymerase epsilon, is an enzyme involved in DNA replication and repair. Select POLE mutations lead to ultra-high m

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR