NM_020975.6:c.2689C>T (p.Arg897Ter) is a nonsense variant in exon 15 of 20 in the RET gene. Loss-of-function variants in RET are an established cause of Hirschsprung disease.1 Nonsense-mediated decay is predicted as the premature termination codon at Arg897 is located >50 nucleotides upstream of the last exon-exon junction. This variant qualifies for PVS1 at very strong strength under the ClinGen SVI PVS1 decision tree (PMC6185798).2 The variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada population databases, supporting PM2 at moderate strength.3 ClinVar reports this variant as Pathogenic from two clinical testing laboratories (GeneDx and Labcorp/Invitae), supporting PP5 at supporting strength.4 Applying the generic ACMG/AMP 2015 final classification combination rules: one very strong criterion (PVS1), one moderate criterion (PM2), and one supporting criterion (PP5) combine to support a Pathogenic classification.5
RET
Final classification
Pathogenic
RET c.2689C>T · p.Arg897Ter
RET
NM_020975.6:c.2689C>T (p.Arg897Ter) is a nonsense variant in exon 15 of 20 in the RET gene. Loss-of-function variants in RET are an established cause of Hirschsprung disease.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 moderate, PP5 supporting; combination = 1 very strong + 1 moderate + 1 supporting, which maps to Pathogenic.
Classification rationale
PVS1PM2PP5
Pathogenic
RET c.2689C>T
PVS1 + PM2 + PP5
→
Pathogenic
1
pvs1_variant_assessmentpvs1_gene_context
5
generic_acmg_combination_rules
Gene diagram
· NM_020975.6 · variants mapped to exon structure
RET
NM_020975.6
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 15 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_020975.6:c.2689C>T is a nonsense variant introducing a premature termination codon at p.Arg897Ter in exon 15 of 20. Nonsense-mediated decay is predicted. RET loss-of-function is an established germline disease mechanism for Hirschsprung disease. Under the ClinGen SVI PVS1 decision tree (PMC6185798), this null variant in a gene with confirmed LoF mechanism qualifies for PVS1 at very strong strength.
Nonsense variant p.Arg897Ter in exon 15 of 20 (NM_020975.6). NMD predicted: PTC located >50 nt upstream of the last exon-exon junction.RET loss-of-function is an established germline disease mechanismsupported by literature linking RET LoF variants to Hirschsprung disease (HSCR).
✓
PM2
moderate
Pathogenic
This variant is absent from gnomAD v2.1, gnomAD v4.1, and gnomAD-Canada v1.0, indicating it is not a common benign polymorphism in population databases (allele frequency <0.1%).
Absent from gnomAD v2.1 (exomes).Absent from gnomAD v4.1 (exomes).Absent from gnomAD-Canada v1.0 (genomes).
✓
PP5
supporting
Pathogenic
ClinVar classifies this variant as Pathogenic based on submissions from two clinical testing laboratories (GeneDx and Labcorp/Invitae). While the review status is 'criteria provided, single submitter' and the cited PMIDs do not specifically name this variant, the Pathogenic classification from multiple reputable clinical laboratories is consistent with the predicted impact of a nonsense variant in a gene with established loss-of-function disease mechanism.
ClinVar variation ID 405536: Pathogenic classification from 2 clinical laboratories (GeneDx SCV002498866Labcorp/Invitae SCV000543814).Both submissions are from clinical testing laboratories with criteria provided.
Assessed · not applied
Pathogenic
PS2
No de novo occurrence data are available for this variant in the case materials or literature reviewed.
PS3
No variant-specific functional studies were identified for NM_020975.6:c.2689C>T (p.Arg897Ter).
PS4
No case-control data comparing the prevalence of this variant in affected individuals versus controls are available.
PM1
While the variant truncates RET within the tyrosine kinase domain 1 (aa 724-1016), a well-characterized functional domain, this truncation effect is already fully captured by PVS1.
PM6
No de novo data are available for this variant; parental testing results have not been provided.
PP1
No co-segregation data are available for this variant.
PP4
No patient phenotype or family history information has been provided in the case materials for evaluation.
Benign
BA1
The variant is absent from gnomAD v2.1 and v4.1, far below the BA1 threshold of >1% allele frequency in population databases.
BS1
The variant is absent from gnomAD, far below the BS1 threshold of >0.3% allele frequency.
BS2
No evidence is available that this variant has been observed in healthy adult controls with full penetrance expected.
BS3
No well-established functional studies demonstrate a benign effect for this variant.
BS4
No non-segregation data are available for this variant.
BP2
No evidence that this variant has been observed in trans with a pathogenic variant in a recessive disorder.
BP5
No evidence that this variant has been observed in a case with an alternate molecular basis for disease.
BP6
ClinVar classifies this variant as Pathogenic, not Benign.
N/A · 10
PS1 · PM3 · PM4 · PM5 · PP2 · PP3 · BP1 · BP3 · BP4 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
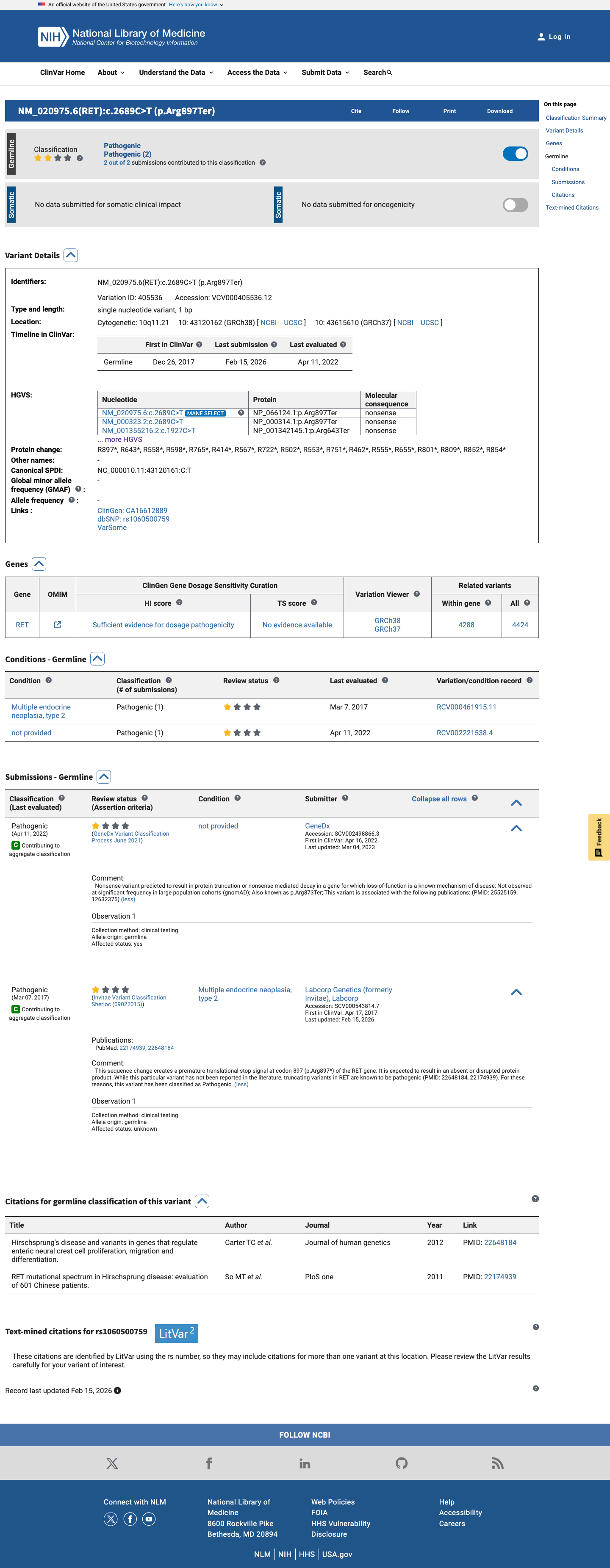
ClinVar
This variant has been reported in ClinVar as Pathogenic (2 clinical laboratories). (ClinVarID = 405536)
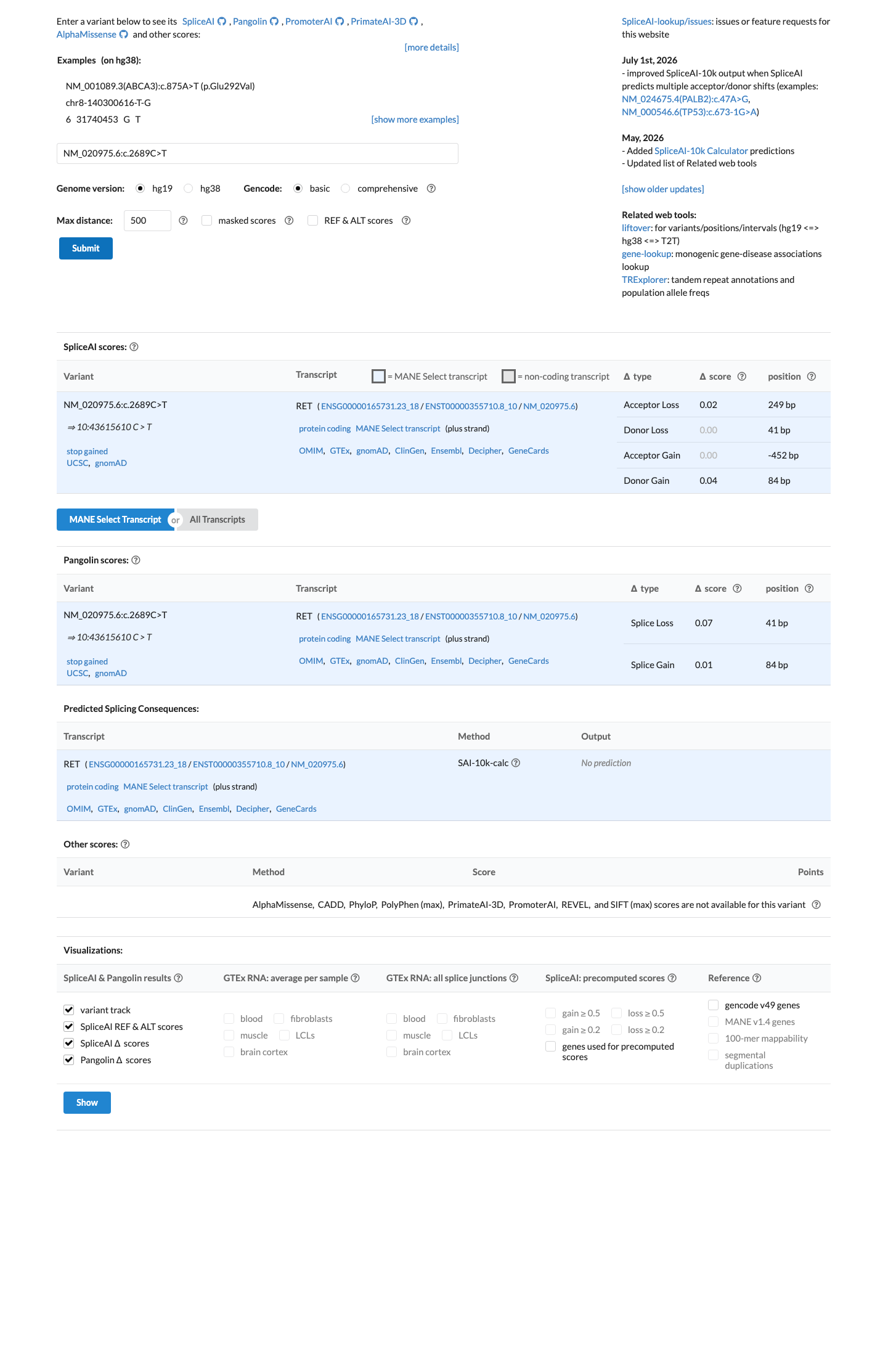
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.04). BayesDel score = 0.66.
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.
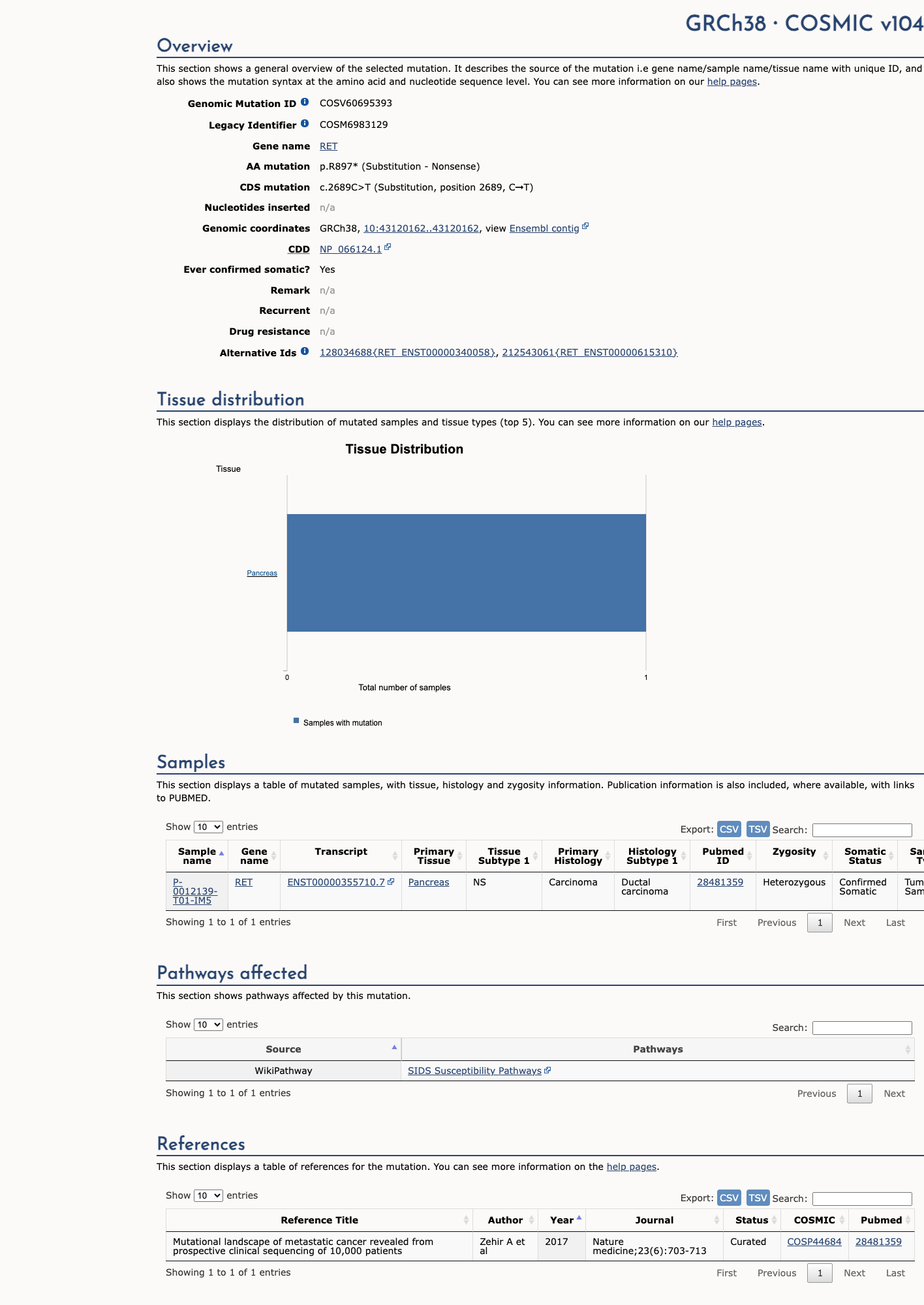
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV60695393, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
22174939 ↗
RET mutational spectrum in Hirschsprung disease: evaluation of 601 Chinese patients.
CLINVAR
22648184 ↗
Hirschsprung's disease and variants in genes that regulate enteric neural crest cell proliferation, migration and differentiation.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
26389258 ↗
Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version.
CLINVAR
26389271 ↗
Genetics of Endocrine and Neuroendocrine Neoplasias (PDQ®): Health Professional Version.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR