NM_000465.4:c.1325C>T (p.Pro442Leu) is a missense variant in BARD1 present at extremely low frequency in population databases (gnomAD v2.1 AF=0.00119%; v4.1 AF=0.00236%), meeting PM2 at supporting level.1 Multiple in silico tools consistently predict a benign effect: REVEL score 0.208, BayesDel score -0.244, and SpliceAI max delta 0.07, meeting BP4 at supporting level.2 BARD1 is a tumor suppressor gene where primarily loss-of-function variants cause disease; this missense variant meets BP1 at supporting level.3 No functional studies, case-control data, de novo observations, co-segregation data, or pathogenic same-residue comparators exist for this variant. ClinVar classifies it as a Variant of Uncertain Significance (9 clinical laboratories).4 Two supporting benign criteria (BP1 and BP4) are met with no moderate or strong criteria on either side. Per generic ACMG/AMP 2015 combination rules, two supporting benign criteria warrant a classification of Likely Benign.5
BARD1
Final classification
Likely Benign
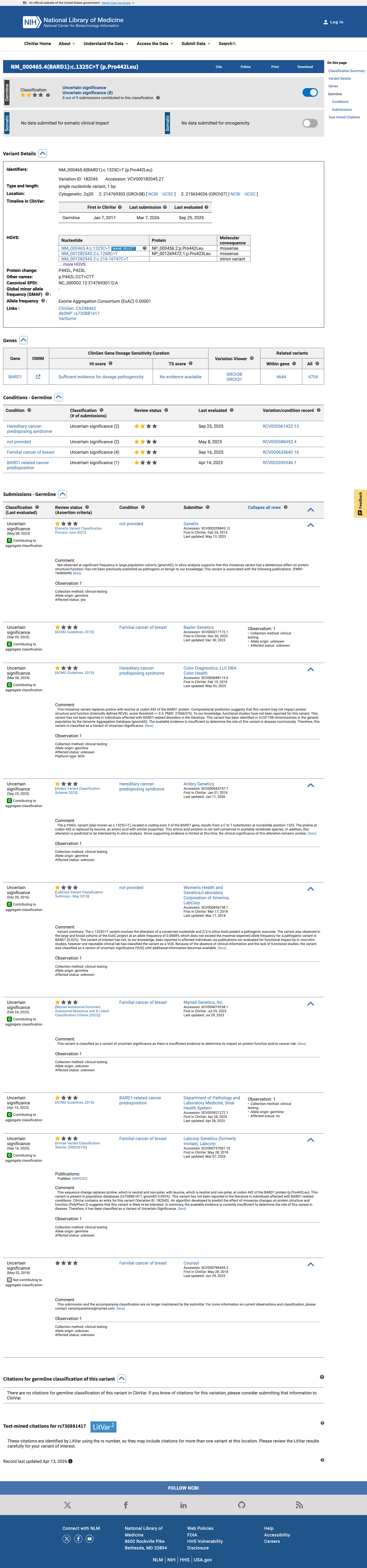
BARD1 c.1325C>T · p.Pro442Leu
BARD1
NM_000465.4:c.1325C>T (p.Pro442Leu) is a missense variant in BARD1 present at extremely low frequency in population databases (gnomAD v2.1 AF=0.00119%; v4.1 AF=0.00236%), meeting PM2 at supporting level.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP1 supporting benign, BP4 supporting benign; combination = 1 supporting + 2 supporting benign, which maps to Likely Benign.
Classification rationale
PM2
BP1BP4
Likely Benign
BARD1 c.1325C>T
PM2 + BP1 + BP4
→
Likely Benign
2
revelbayesdelspliceai ↗
3
pvs1_gene_context
4
clinvar ↗pm5_candidates
5
generic_acmg_combination_rules
Gene diagram
· NM_000465.4 · variants mapped to exon structure
BARD1
NM_000465.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 19 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
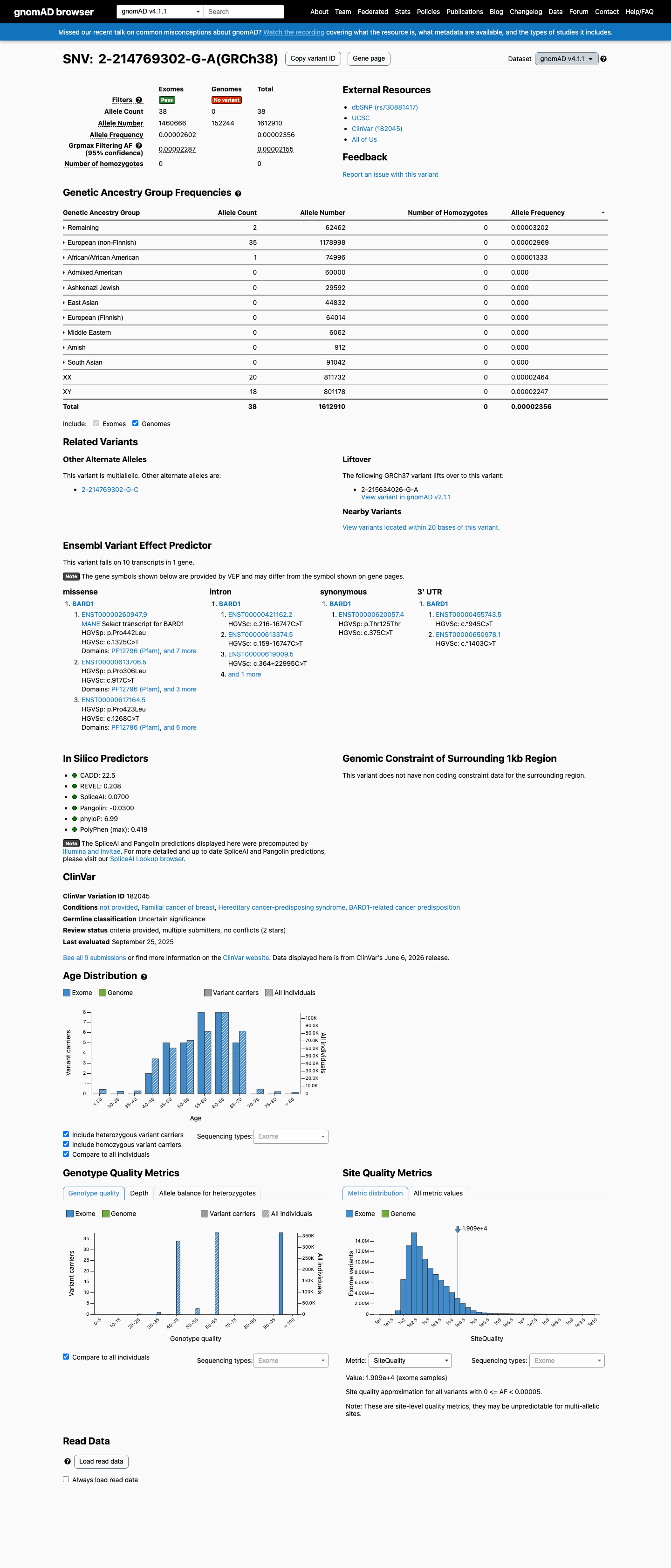
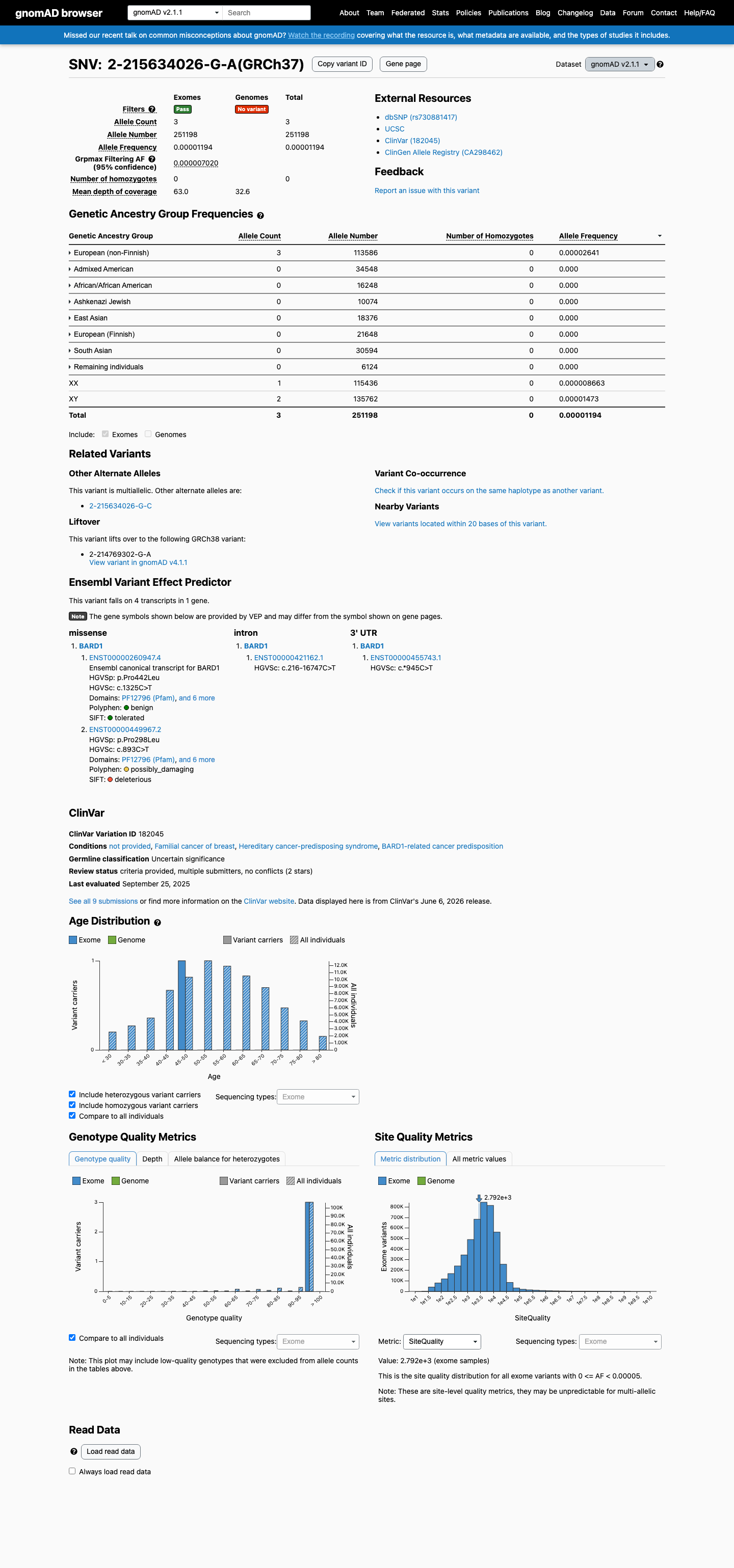
This variant is present at extremely low frequency in population databases. gnomAD v2.1: 3/251,198 alleles (AF=0.00119%); gnomAD v4.1: 38/1,612,910 alleles (AF=0.00236%); absent from gnomAD-Canada. All allele frequencies are well below the 0.1% PM2 threshold. No homozygotes observed.
gnomAD v2.1 AF=0.00119% (3/251198 alleles0 homozygotes)
✓
BP1
supporting
Benign
BARD1 is a tumor suppressor gene where primarily loss-of-function (truncating, nonsense, splice-disrupting) variants are known to cause disease. NM_000465.4:c.1325C>T is a missense variant (p.Pro442Leu), and missense variants are not the established disease mechanism for BARD1.
BARD1 germline LoF mechanism is supported by disease literature (PVS1 gene context). Missense variants are not the primary pathogenic mechanism.
✓
BP4
supporting
Benign
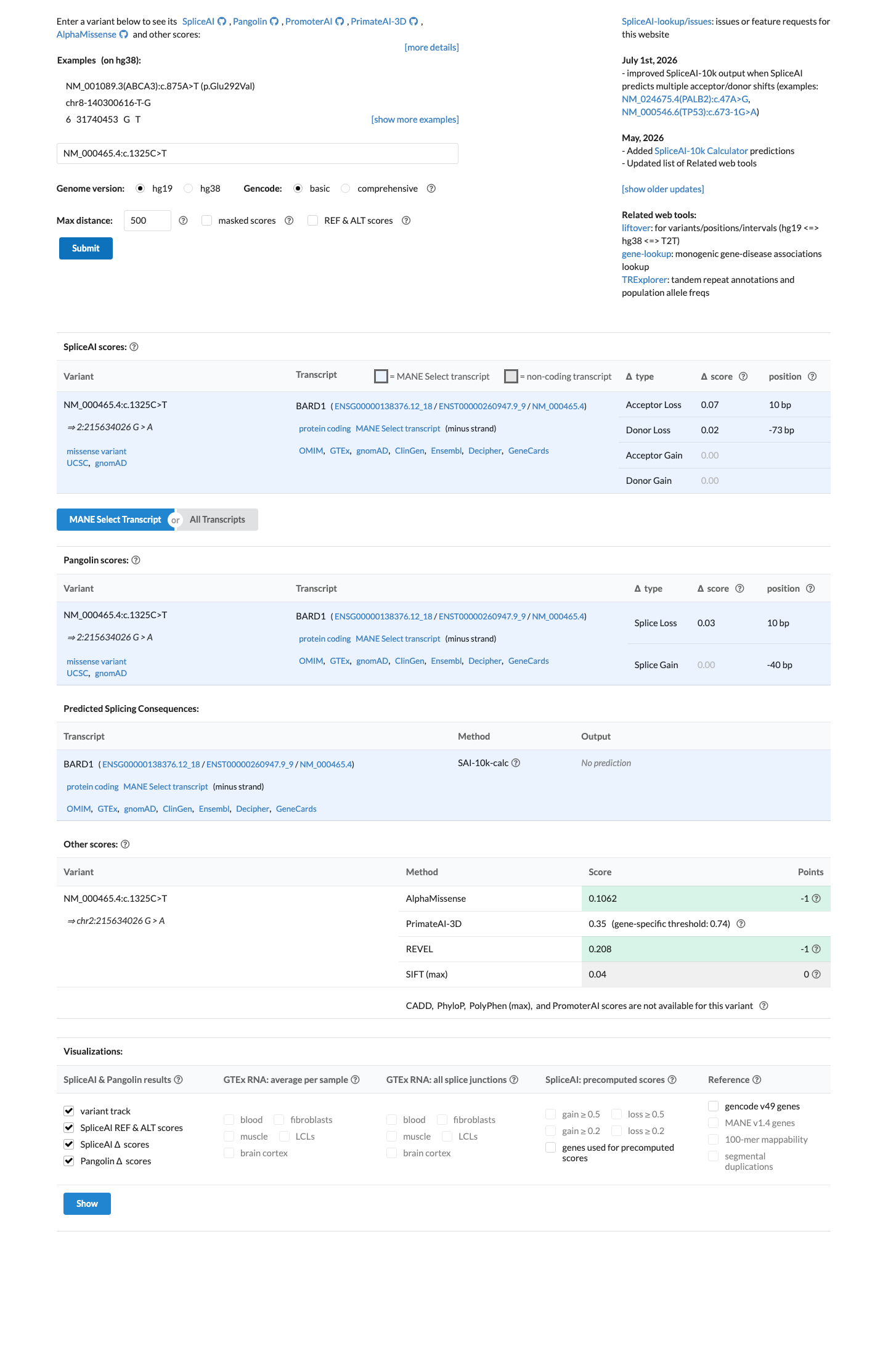
Multiple lines of computational evidence support a benign effect: REVEL score 0.208 (well below the pathogenic threshold of ~0.5), BayesDel score -0.243647 (negative/benign), and SpliceAI max delta score 0.07 (no predicted splice impact). These independent in silico tools consistently suggest this missense change is tolerated.
REVEL=0.208 (benign)BayesDel=-0.244 (benign)SpliceAI max delta=0.07 (no splice impact). ClinVar submitter Ambry Genetics notes the alteration is predicted to be tolerated by in silico analysis.
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant at amino acid position 442 (Pro442) with a different substitution has been identified in ClinVar or the reviewed literature.
PS2
No de novo data are available for this variant.
PS3
No functional studies have directly tested BARD1 Pro442Leu or a systematically characterized range that includes position 442.
PS4
No case-control studies or sufficient enrichment data in affected individuals are available for this variant.
PM1
The variant (p.Pro442Leu) does not lie in a statistically significant mutational hotspot as assessed by cancerhotspots.org.
PM6
No de novo observation has been reported for this variant.
PP1
No co-segregation data are available for this variant.
PP2
BARD1 is a tumor suppressor gene where loss-of-function (truncating, nonsense, splice) variants are the established disease mechanism.
PP3
Multiple in silico tools predict a benign effect for this variant: REVEL score 0.208 (well below the pathogenic threshold of ~0.5), BayesDel score -0.243647 (negative, benign), and SpliceAI max delta 0.07 (no predicted splice impact).
PP4
No patient phenotype or specific clinical data are available for the individuals carrying this variant.
PP5
This variant is classified as Uncertain Significance (VUS) in ClinVar by 9 clinical laboratories, not Pathogenic or Likely Pathogenic.
Benign
BA1
The allele frequency of this variant in gnomAD v2.1 (0.00119%) and v4.1 (0.00236%) is well below the 1% BA1 threshold.
BS1
The allele frequency of this variant (gnomAD v2.1: 0.00119%, v4.1: 0.00236%) is below the 0.3% BS1 threshold.
BS2
No evidence is available that this variant has been observed in healthy adults with full penetrance expected at an early age.
BS3
No functional studies demonstrating a neutral or benign effect exist for this variant.
BS4
No co-segregation data demonstrating lack of segregation with disease are available.
BP2
No evidence that this variant has been observed in trans with a known pathogenic variant in BARD1.
BP5
No observation of this variant in a case with an alternate molecular basis for disease has been reported.
BP6
ClinVar classifies this variant as Uncertain Significance, not Benign or Likely Benign.
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.35599e-05; MAF= 0.00236%, 38/1612910 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 3.20195e-05; MAF= 0.00320%, 2/62462 alleles, homozygotes = 0); grpmax FAF= 2.155e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 1.19428e-05; MAF= 0.00119%, 3/251198 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 2.64117e-05; MAF= 0.00264%, 3/113586 alleles, homozygotes = 0); grpmax FAF= 7.02e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0024%
· 38 / 1,612,910
0 hom · FAF 0.0022%
0 hom · FAF 0.0022%
Remaining individuals 2 / 62,462 |
0.0032% |
European (non-Finnish) 35 / 1,178,998 |
0.003% |
African/African American 1 / 74,996 |
0.0013% |
+ 7 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0012%
· 3 / 251,198
0 hom · FAF 0.0007%
0 hom · FAF 0.0007%
European (non-Finnish) 3 / 113,586 |
0.0026% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (9 clinical laboratories). (ClinVarID = 182045)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.07). REVEL score = 0.208. BayesDel score = -0.243647.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. BARD1, a tumor suppressor involved in the DNA damage response, is altered by mutation in breast and ovarian cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
34242744 ↗
Customizing local and systemic therapies for women with early breast cancer: the St. Gallen International Consensus Guidelines for treatment of early breast cancer 2021.
CLINVAR
24366402 ↗
Summaries for patients. Assessing the genetic risk for BRCA-related breast or ovarian cancer in women: recommendations from the U.S. Preventive Services Task Force.
CLINVAR
26389258 ↗
Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version.
CLINVAR
35802134 ↗
ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR