Classification rationale
PM1PM2PM5PP5
Likely Pathogenic
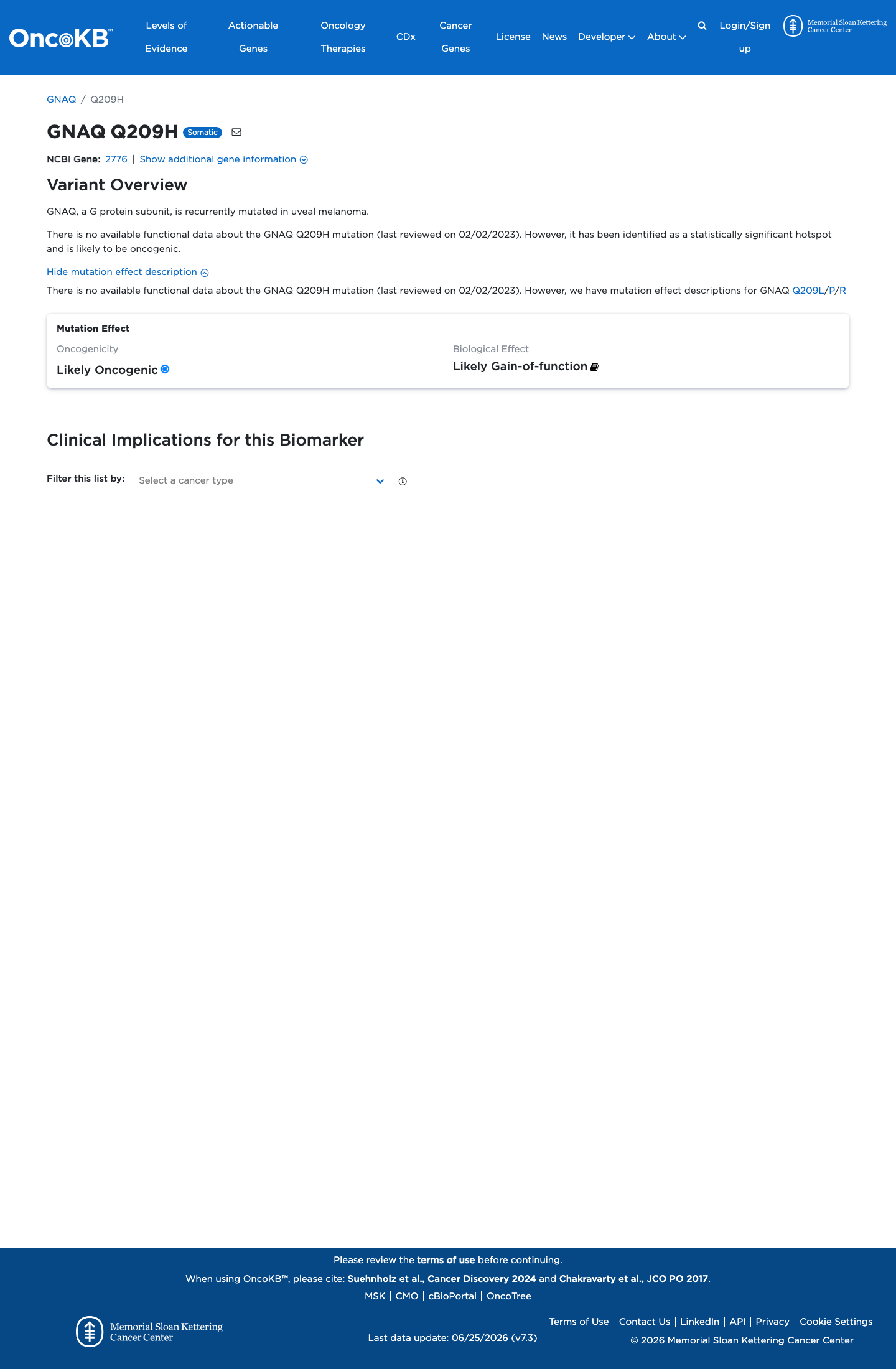
GNAQ c.627A>C
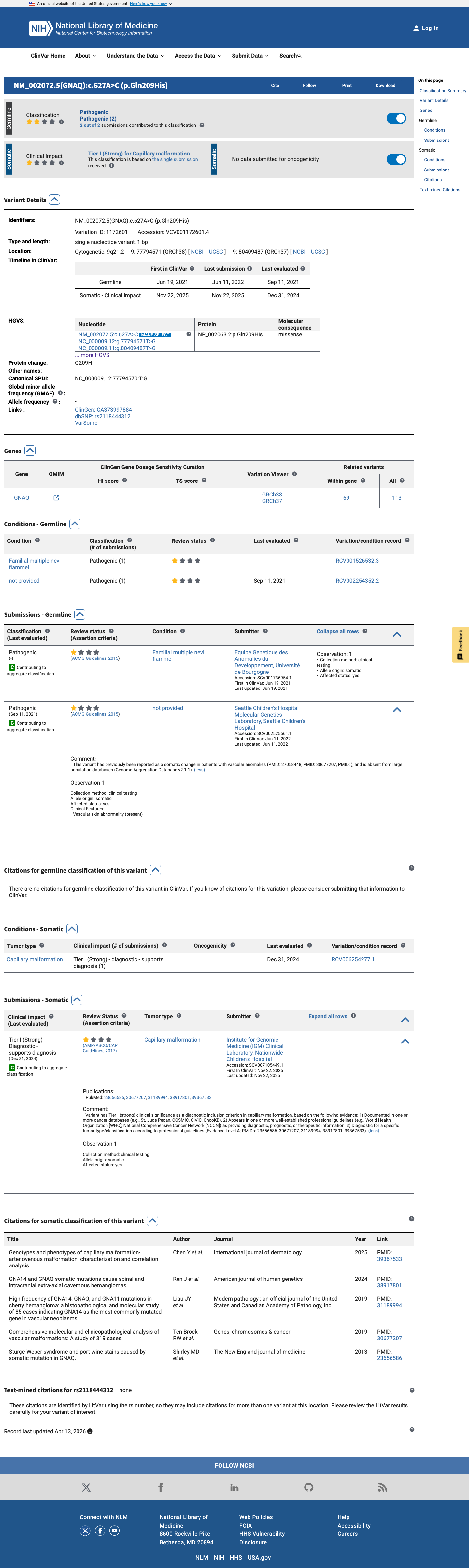
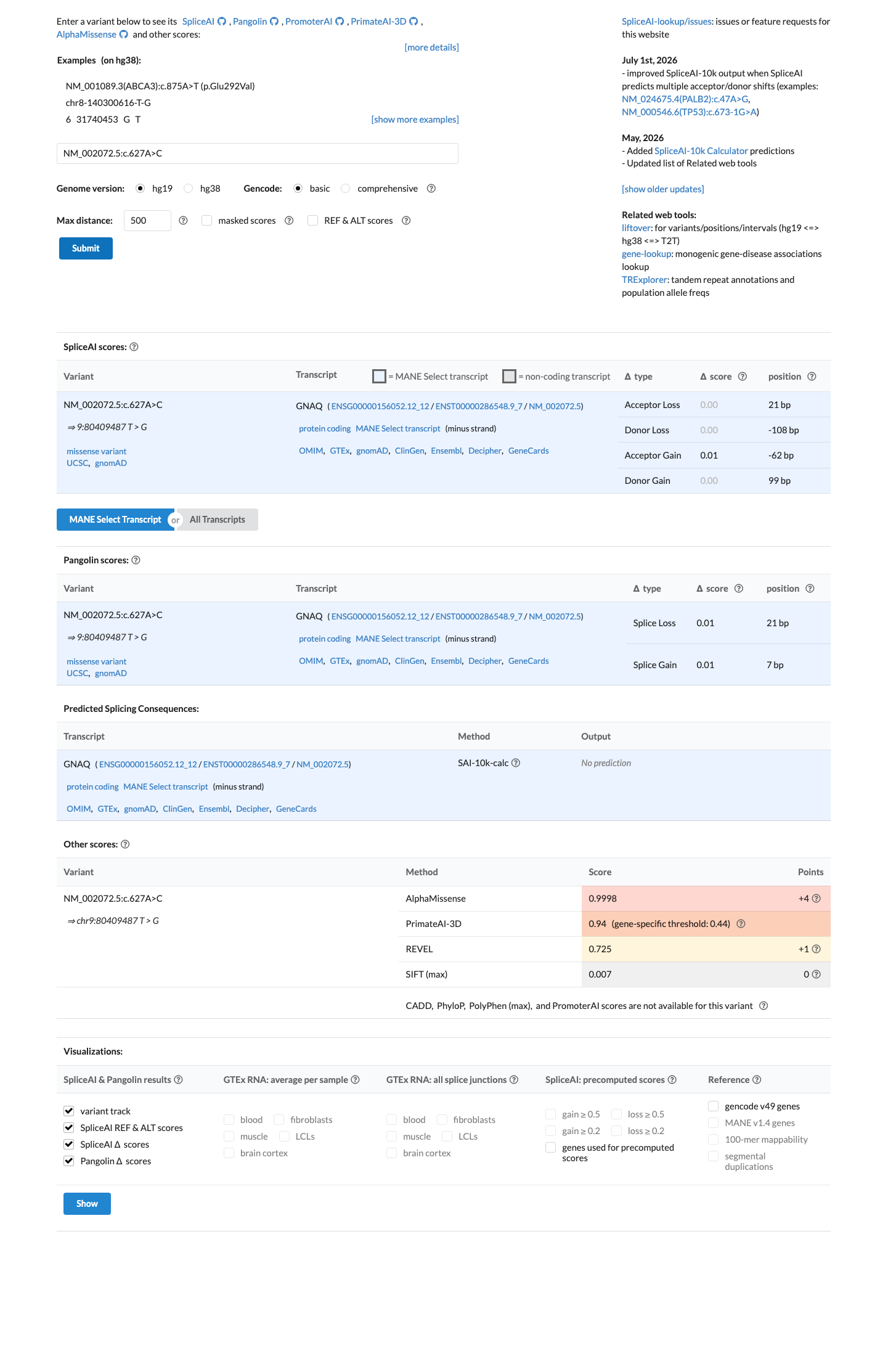
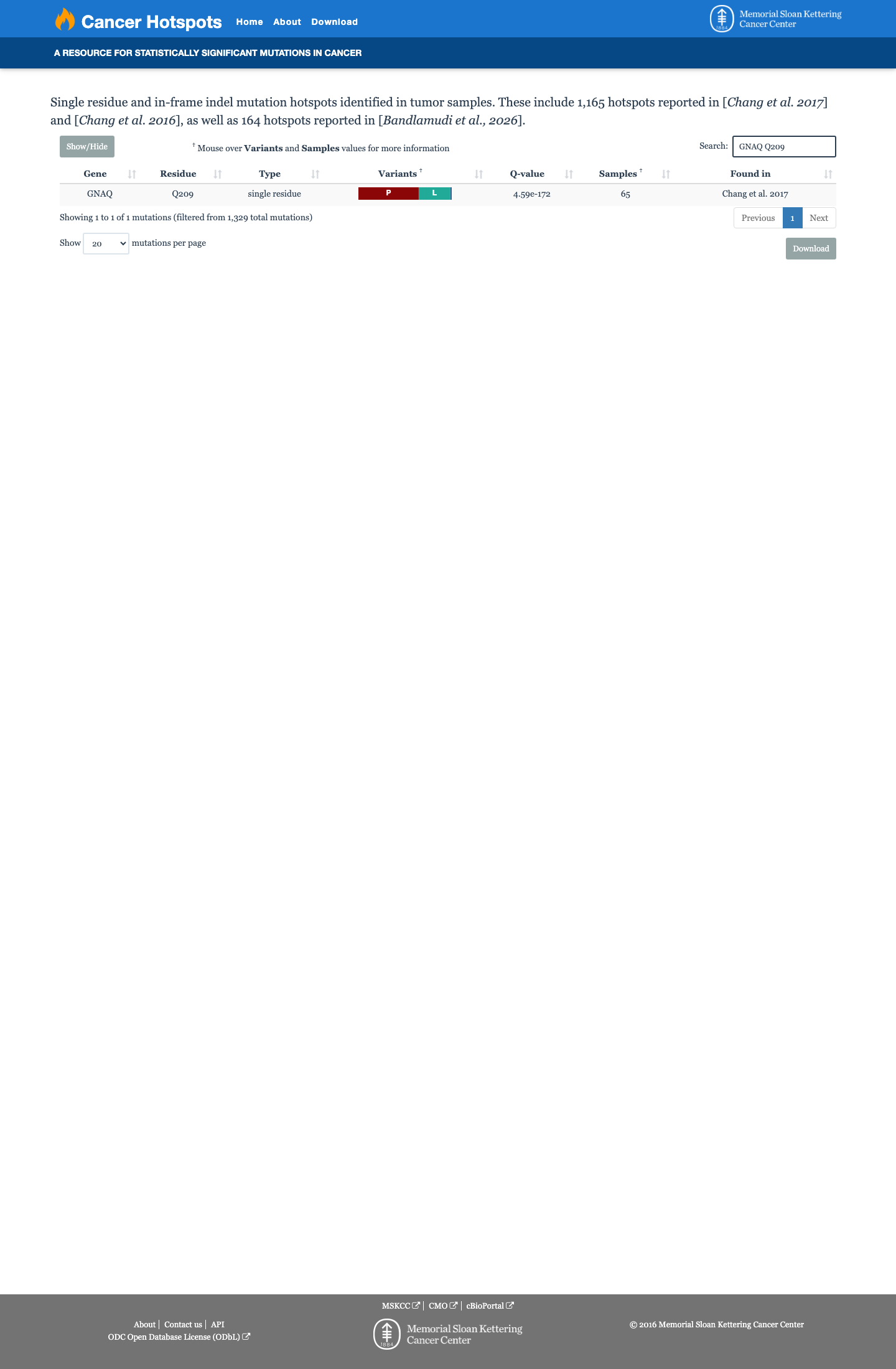
The variant is located at codon 209 within the GTPase domain of GNAQ, a well-established functional hotspot (PM1).1 The variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada (PM2).2 A different pathogenic missense change at the same codon, p.Gln209Leu, has been established as a gain-of-function activating mutation in functional studies (PM5).3 This variant has been reported as Pathogenic in ClinVar by two clinical laboratories (PP5).4 Three moderate criteria (PM1, PM2, PM5) and one supporting criterion (PP5) are met, reaching the Likely Pathogenic threshold per ACMG/AMP 2015 generic combination rules (3 moderate criteria).5
PM1 + PM2 + PM5 + PP5
→
Likely Pathogenic
5
generic_acmg_combination_rulesPMID:25741868 ↗