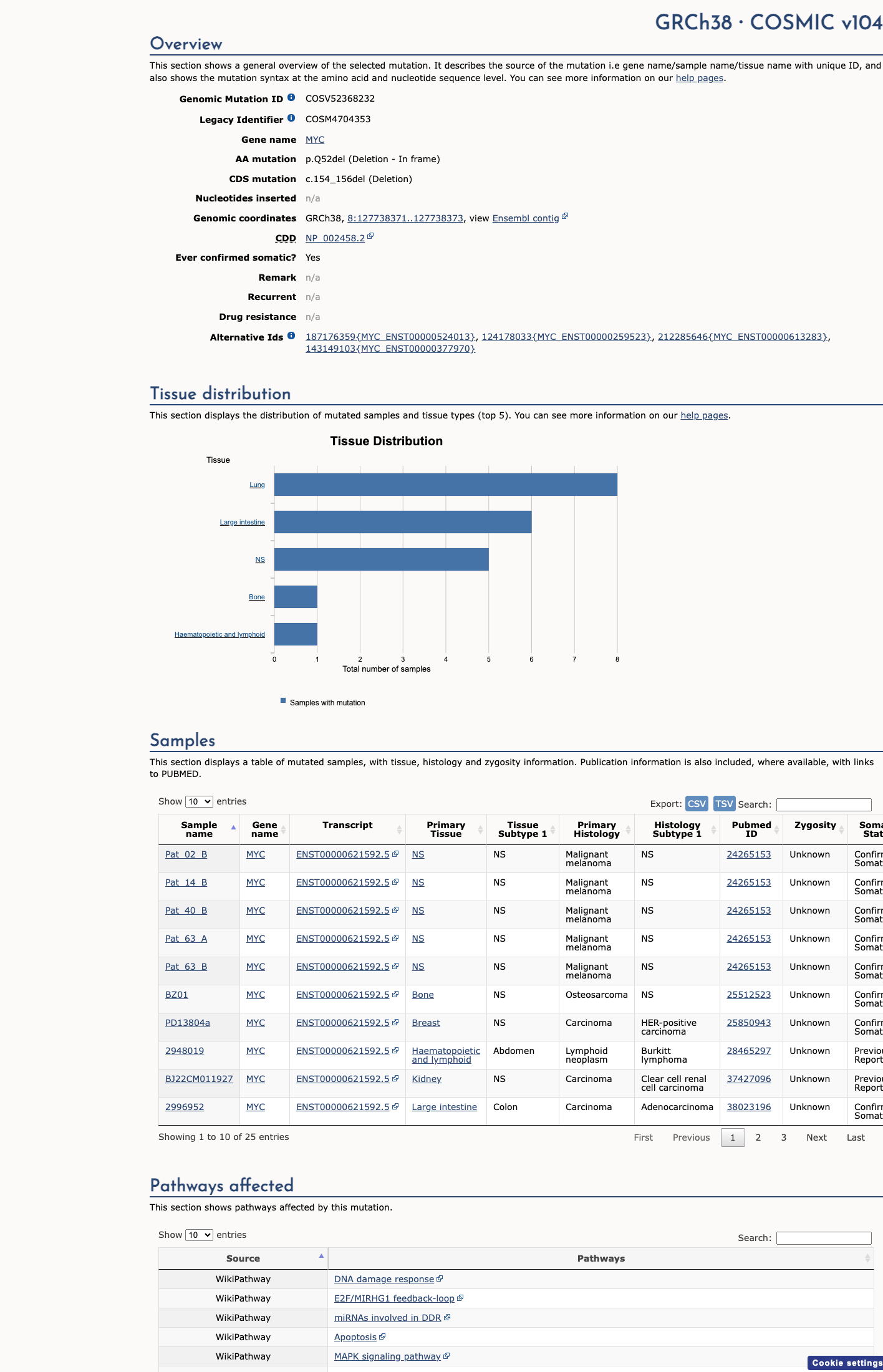
NM_002467.5:c.154_156del (p.Gln52del) is an in-frame deletion of a single amino acid in exon 2 of MYC, encoding the MYC proto-oncogene transcription factor. This variant deletes glutamine at position 52, located within the well-characterized MYC Box I (MB1) transactivation domain (residues 44–63). PM1 (supporting) is applied.1 The variant is extremely rare in population databases: gnomAD v2.1 allele frequency = 0.0016% (4/249,036 alleles) and gnomAD v4.1 allele frequency = 0.00093% (15/1,612,048 alleles), with no homozygotes observed. PM2 (moderate) is applied.2 As an in-frame deletion in a non-repeat region resulting in a protein length change (loss of one amino acid), PM4 (moderate) is applied. PVS1 is not met: this in-frame deletion does not qualify as a predicted null variant under the ClinGen SVI PVS1 decision framework (variant bucket: other). Nonsense-mediated decay is not expected.3 PS3 is not met: no well-established functional studies were identified for this variant. Literature review yielded zero PMIDs with variant-specific data. OncoKB reports unknown oncogenic effect.4 This variant has been observed 25 times in somatic cancers (COSMIC COSV52368232) but has not been reported in ClinVar. No germline disease associations, de novo events, cosegregation data, or patient phenotypes are available.5 SpliceAI predicts no splicing impact (max delta score = 0.00). In silico pathogenicity tools (REVEL, BayesDel) are not applicable to deletion variants.6
MYC
Final classification
VUS
MYC c.154_156del · p.Gln52del
MYC
NM_002467.5:c.154_156del (p.Gln52del) is an in-frame deletion of a single amino acid in exon 2 of MYC, encoding the MYC proto-oncogene transcription factor.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 supporting, PM2 moderate, PM4 moderate; combination = 2 moderate + 1 supporting, which maps to VUS.
Classification rationale
PM1PM2PM4
VUS
MYC c.154_156del
PM1 + PM2 + PM4
→
VUS
Gene diagram
· NM_002467.5 · variants mapped to exon structure
MYC
NM_002467.5
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 20 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
supporting
Pathogenic
The variant deletes glutamine at position 52, which lies within MYC Box I (MB1, residues 44–63), a well-characterized transactivation domain critical for MYC-mediated transcription and transformation. The in-frame deletion removes a residue from this critical functional domain. The residue-level cancerhotspots.org analysis did not identify a statistically significant hotspot at this position.
MYC Box I (MB1) spans residues 44–63 and is a well-characterized transactivation domainp.Gln52del removes a single residue from within MB1Cancerhotspots.org: residue not statistically significant
✓
PM2
moderate
Pathogenic
This variant is absent or at extremely low frequency in population databases. In gnomAD v2.1, the allele frequency is 0.0016% (4/249,036 alleles, no homozygotes). In gnomAD v4.1, the allele frequency is 0.00093% (15/1,612,048 alleles, no homozygotes). Both are well below the 0.1% threshold for PM2. The variant is absent from gnomAD-Canada v1.0.
gnomAD v2.1: AF = 0.0016% (4/249036)0 homozygotes
✓
PM4
moderate
Pathogenic
NM_002467.5:c.154_156del is an in-frame 3-nucleotide deletion resulting in the loss of a single amino acid (p.Gln52del) in a non-repeat region of MYC. This protein length change satisfies the PM4 criterion for in-frame deletions in non-repeat regions.
In-frame deletion of 3 nucleotides (c.154_156del) causing loss of Gln52No evidence that position 52 lies within a repetitive region
Assessed · not applied
Pathogenic
PVS1
NM_002467.5:c.154_156del is an in-frame deletion of a single amino acid (p.Gln52del) and does not fall into the ClinGen SVI PVS1 null-variant buckets (nonsense, frameshift, or canonical ±1,2 splice consensus variants).
PS1
No previously established pathogenic variant causing the same amino acid change (p.Gln52del) was identified.
PS2
No de novo occurrence data are available for this variant.
PS3
No well-established in vitro or in vivo functional studies supporting a damaging effect were identified for NM_002467.5:c.154_156del.
PS4
No case-control or cohort prevalence data comparing affected versus unaffected individuals are available for this variant.
PM6
No de novo occurrence data (assumed or confirmed) are available for this variant.
PP1
No cosegregation data are available for this variant.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No patient phenotype or clinical data are available for this case.
PP5
No reputable source has reported this variant as pathogenic.
Benign
BA1
The allele frequency in population databases is well below the 1% BA1 threshold.
BS1
The allele frequency is well below the 0.3% BS1 threshold for non-VCEP adjudication.
BS2
No homozygotes are observed in gnomAD v2.1 or v4.1.
BS3
No well-established in vitro or in vivo functional studies showing no deleterious effect were identified for this variant.
BS4
No cosegregation or family data are available to evaluate lack of segregation with disease.
BP2
No evidence that this variant has been observed in trans with a known pathogenic variant in a gene associated with a fully penetrant dominant disorder.
BP3
BP3 applies to in-frame deletions or insertions in repetitive regions without known function.
BP4
Multiple lines of computational evidence do not conclusively suggest no impact.
BP5
No data are available regarding an alternate molecular basis for disease in a case harboring this variant.
BP6
No reputable source has reported this variant as benign.
N/A · 5
PM3 · PM5 · PP2 · BP1 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
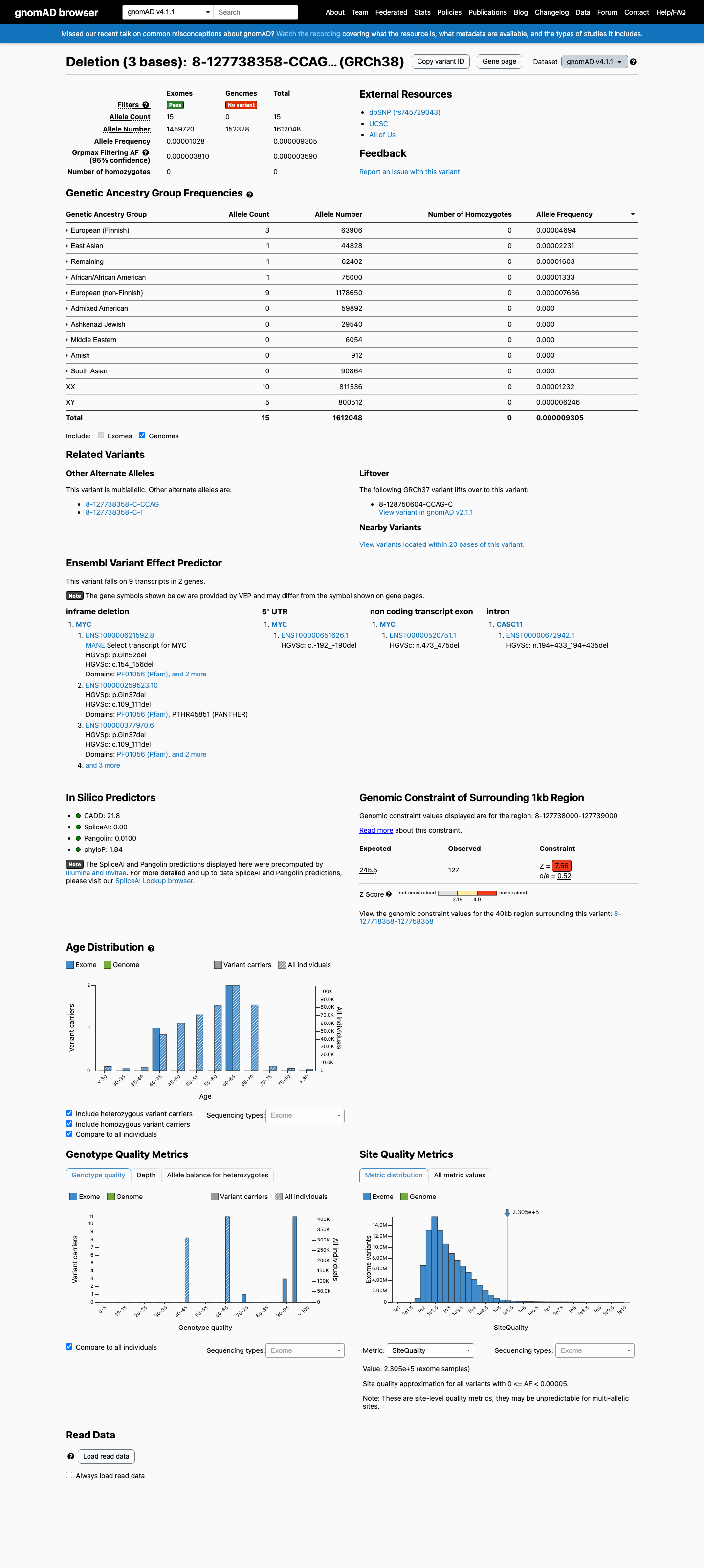
This variant is present in gnomAD v4.1 (AF= 9.30493e-06; MAF= 0.00093%, 15/1612048 alleles, homozygotes = 0) and has highest observed frequency in the European (Finnish) population (AF= 4.69439e-05; MAF= 0.00469%, 3/63906 alleles, homozygotes = 0); grpmax FAF= 3.59e-06.
v2.1
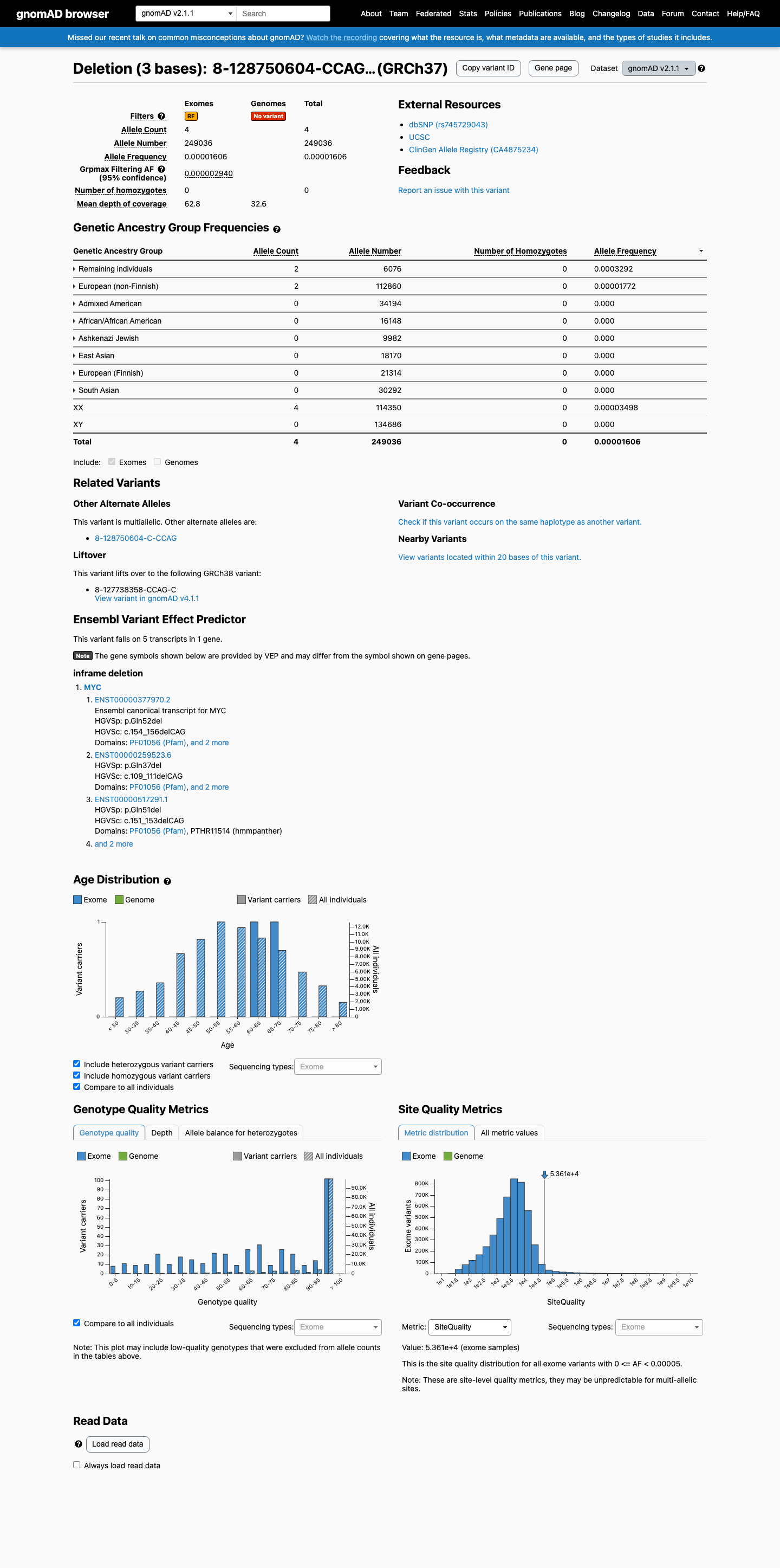
This variant is present in gnomAD v2.1 (AF= 1.60619e-05; MAF= 0.00161%, 4/249036 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.000329164; MAF= 0.03292%, 2/6076 alleles, homozygotes = 0); grpmax FAF= 2.94e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00093%
· 15 / 1,612,048
0 hom · FAF 0.00036%
0 hom · FAF 0.00036%
European (Finnish) 3 / 63,906 |
0.0047% |
East Asian 1 / 44,828 |
0.0022% |
Remaining individuals 1 / 62,402 |
0.0016% |
African/African American 1 / 75,000 |
0.0013% |
European (non-Finnish) 9 / 1,178,650 |
0.00076% |
+ 5 not observed (Admixed American, Amish, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0016%
· 4 / 249,036
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
Remaining individuals 2 / 6,076 |
0.033% |
European (non-Finnish) 2 / 112,860 |
0.0018% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MYC, a transcription factor, is altered by chromosomal rearrangement, amplification and overexpression in a variety of cancer types.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV52368232, n = 25 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links