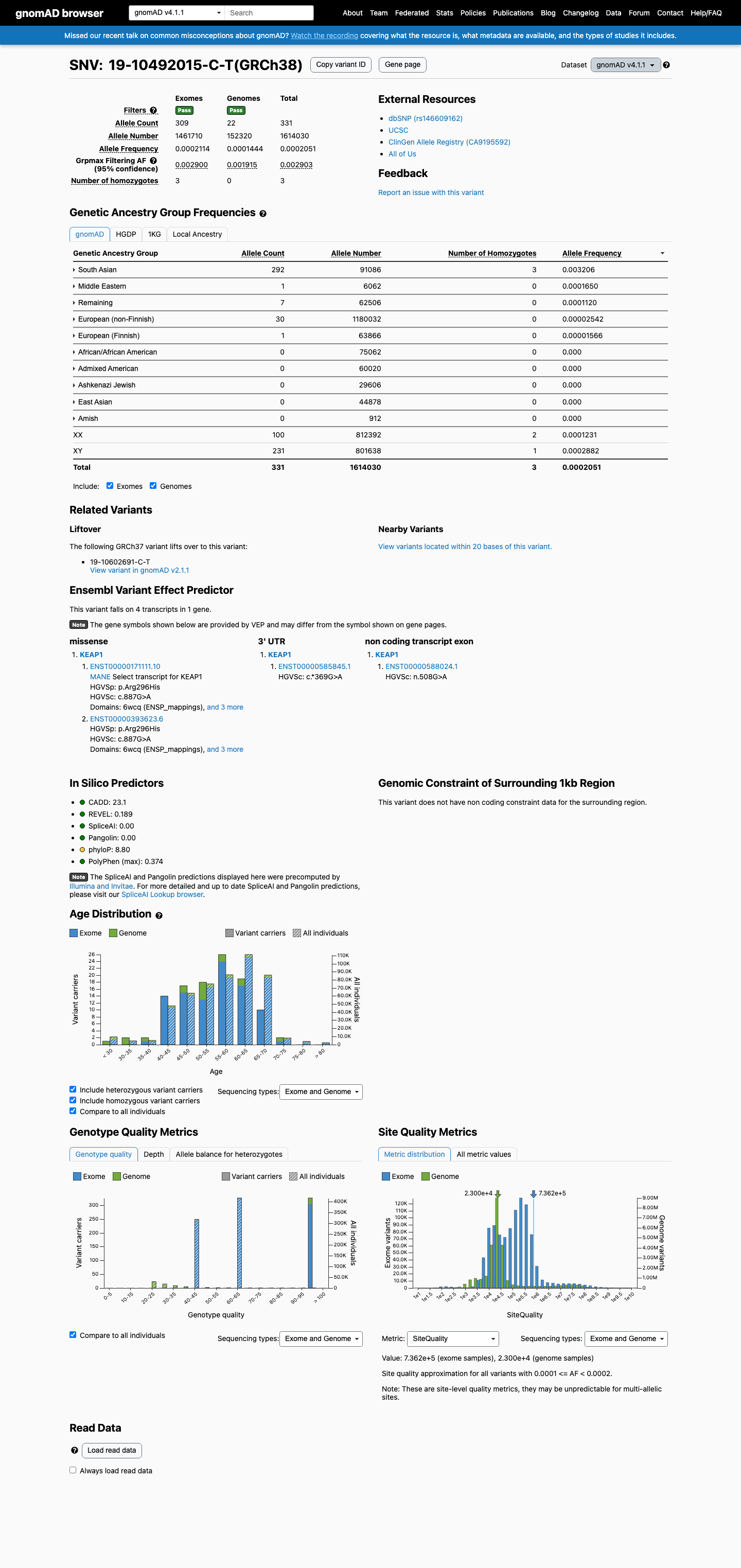
NM_012289.4:c.887G>A (p.Arg296His) is present in the South Asian population at an allele frequency of 0.34% (gnomAD v2.1: 104/30,614 alleles) and 0.32% (gnomAD v4.1: 292/91,086 alleles), exceeding the 0.3% threshold for BS1.1 Three homozygous individuals for c.887G>A are observed in gnomAD v4.1 (2 female, 1 male, all South Asian), which is incompatible with a fully penetrant autosomal dominant disorder such as KEAP1-related familial multinodular goiter (PMID:39373520).2 Multiple in silico tools predict a benign effect: REVEL score 0.189 (below pathogenicity threshold), BayesDel score -0.262 (benign), and SpliceAI max delta 0.00 (no splice impact).3 The variant is absent from ClinVar with no disease-associated submissions, and absent from COSMIC with no somatic cancer reports.4 Computational evidence (BP4) and strong population evidence (BS1, BS2: elevated subpopulation frequency and homozygous observations) collectively support a Benign classification per ACMG/AMP 2015 rules: two strong benign criteria (BS1 + BS2) meet the threshold for Benign.5
KEAP1
Final classification
Benign
KEAP1 c.887G>A · p.Arg296His
KEAP1
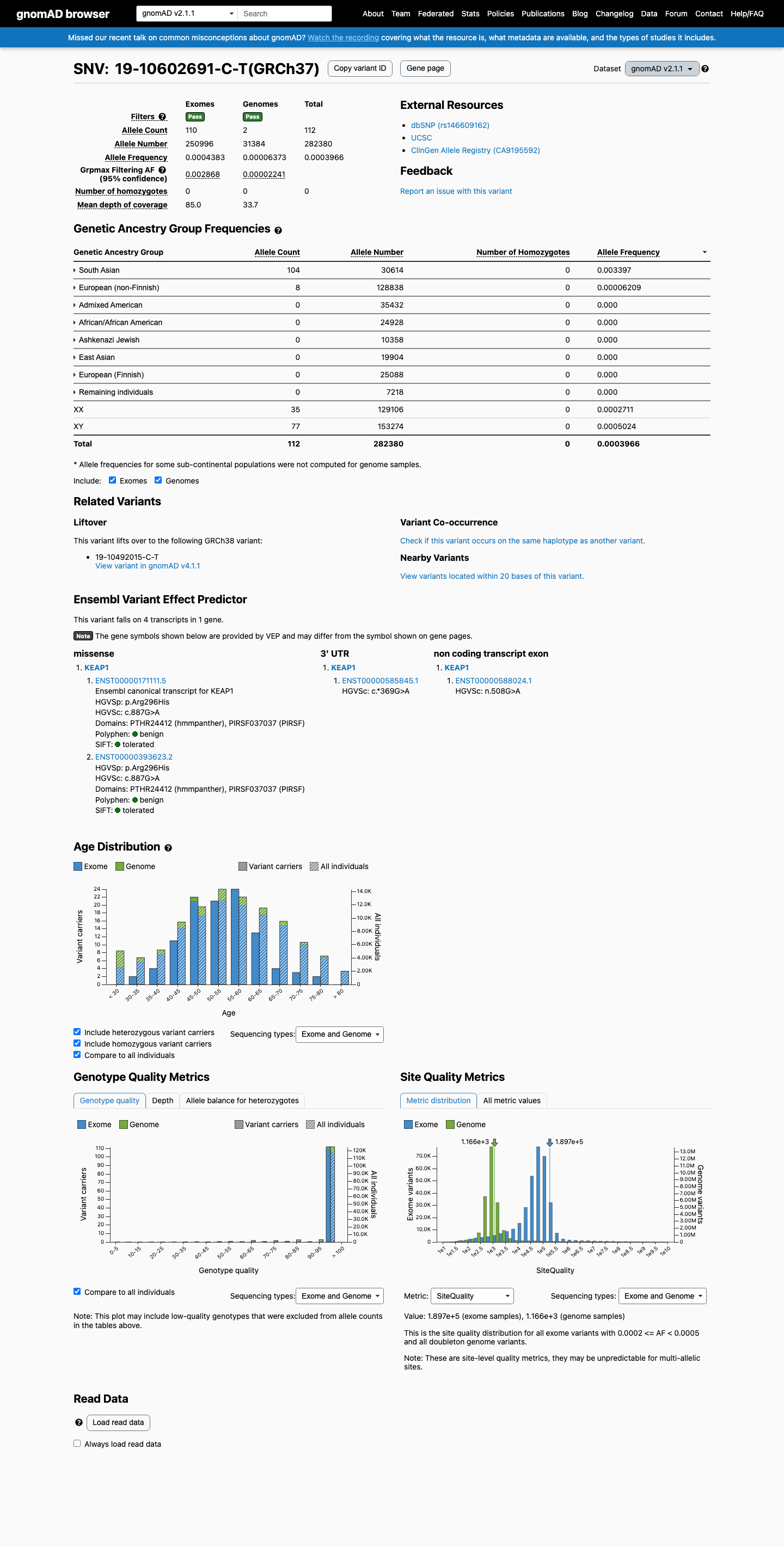
NM_012289.4:c.887G>A (p.Arg296His) is present in the South Asian population at an allele frequency of 0.34% (gnomAD v2.1: 104/30,614 alleles) and 0.32% (gnomAD v4.1: 292/91,086 alleles), exceeding the 0.3% threshold for BS1.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong, BS2 strong, BP4 supporting; combination = 2 strong benign + 1 supporting benign, which maps to Benign.
Classification rationale
BS1BS2BP4
Benign
KEAP1 c.887G>A
BS1 + BS2 + BP4
→
Benign
3
revelbayesdelspliceai ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_012289.4 · variants mapped to exon structure
KEAP1
NM_012289.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 18 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
The South Asian subpopulation allele frequency is 0.34% in gnomAD v2.1 (104/30,614 alleles) and 0.32% in gnomAD v4.1 (292/91,086 alleles), exceeding the 0.3% BS1 threshold. This frequency is incompatible with a high-penetrance pathogenic variant for familial multinodular goiter or other KEAP1-related Mendelian disorder. Additionally, 3 homozygotes are observed in gnomAD v4.1, further confirming the variant is too common in the general population to be pathogenic.
SAS AF 0.34% (v2.1104/30614) and 0.32% (v4.1292/91086) > 0.3% BS1 threshold. 3 homozygotes in gnomAD v4.1.
✓
BS2
strong
Benign
Three homozygotes for NM_012289.4:c.887G>A (p.Arg296His) are observed in gnomAD v4.1 (2 female, 1 male, all within the South Asian population). KEAP1 germline mutations cause autosomal dominant familial multinodular goiter (PMID:39373520). Observation of homozygous individuals in a population database is incompatible with a fully penetrant autosomal dominant disorder, strongly supporting a benign interpretation.
3 homozygotes in gnomAD v4.1 (2 SAS_XX females1 SAS_XY male). Incompatible with autosomal dominant fully penetrant disorder.
✓
BP4
supporting
Benign
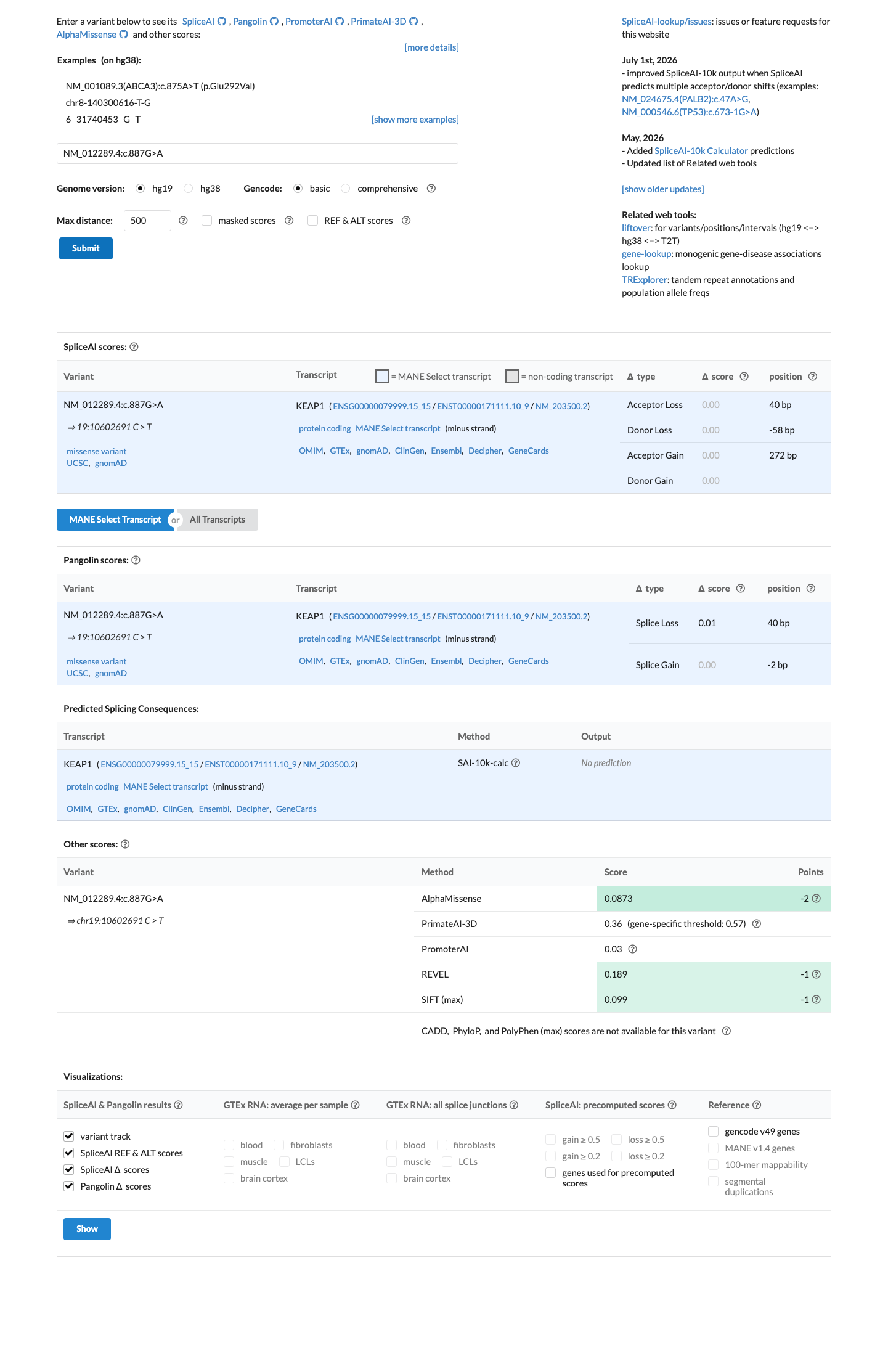
Multiple lines of computational evidence predict a benign effect. REVEL score 0.189 (below 0.5 pathogenicity threshold), BayesDel score -0.262 (negative, predicting benign), and SpliceAI max delta score 0.00 (predicting no splice impact). Three independent in silico tools uniformly support a neutral/benign interpretation.
REVEL 0.189BayesDel -0.262SpliceAI max delta 0.00 — all predict benign/no impact.
Assessed · not applied
Pathogenic
PS2
No de novo occurrence has been reported for NM_012289.4:c.887G>A in any publication or clinical database.
PS3
No functional studies have tested NM_012289.4:c.887G>A (p.Arg296His) directly, nor is residue 296 encompassed by a systematically characterized range (e.g., saturation mutagenesis, CRISPR tiling screen, systematic truncation series) in the published literature.
PS4
No case-control or cohort data comparing variant prevalence in affected versus general population is available.
PM1
Residue 296 (p.Arg296) lies within the KEAP1 IVR (intervening region, aa 180-314), which contains redox-sensitive cysteine residues.
PM2
Overall gnomAD allele frequency (0.04% v2.1, 0.021% v4.1) is below the 0.1% PM2 threshold.
PM6
No de novo occurrence data for NM_012289.4:c.887G>A is available.
PP1
No co-segregation data is available for this variant.
PP2
PP2 requires a low rate of benign missense variation in the gene and missense variants as a common disease mechanism.
PP3
Multiple lines of computational evidence predict a benign effect.
PP4
No patient phenotype or family history data is available for this case.
PP5
No reputable source has reported NM_012289.4:c.887G>A as pathogenic.
Benign
BA1
The highest subpopulation allele frequency is 0.34% in South Asians (gnomAD v2.1).
BS3
No well-established in vitro or in vivo functional studies have tested NM_012289.4:c.887G>A for damaging effect.
BS4
No segregation data is available to demonstrate lack of co-segregation with disease in affected families.
BP1
BP1 applies when a missense variant occurs in a gene for which primarily truncating variants cause disease.
BP2
No data available on whether this variant has been observed in trans with a pathogenic KEAP1 variant.
BP5
No data available demonstrating that this variant has been observed in a case with an alternate molecular basis for disease.
BP6
No reputable source has reported NM_012289.4:c.887G>A as benign.
N/A · 4
PVS1 · PS1 · PM5 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000205077; MAF= 0.02051%, 331/1614030 alleles, homozygotes = 3) and has highest observed frequency in the South Asian population (AF= 0.00320576; MAF= 0.32058%, 292/91086 alleles, homozygotes = 3); grpmax FAF= 0.00290293.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000396629; MAF= 0.03966%, 112/282380 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.00339714; MAF= 0.33971%, 104/30614 alleles, homozygotes = 0); grpmax FAF= 0.00286821.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0002714146129627619, 5/18422 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.021%
· 331 / 1,614,030
3 hom · FAF 0.29%
3 hom · FAF 0.29%
South Asian 292 / 91,086 |
0.32% 3 hom |
Middle Eastern 1 / 6,062 |
0.016% |
Remaining individuals 7 / 62,506 |
0.011% |
European (non-Finnish) 30 / 1,180,032 |
0.0025% |
European (Finnish) 1 / 63,866 |
0.0016% |
+ 5 not observed (Admixed American, Amish, East Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.04%
· 112 / 282,380
0 hom · FAF 0.29%
0 hom · FAF 0.29%
South Asian 104 / 30,614 |
0.34% |
European (non-Finnish) 8 / 128,838 |
0.0062% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
0.027%
· 5 / 18,422
0 hom · FAF 0.1%
0 hom · FAF 0.1%
South Asian 4 / 1,362 |
0.29% |
Remaining individuals 1 / 1,138 |
0.088% |
+ 7 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, European (non-Finnish))

In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.189. BayesDel score = -0.262097.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. KEAP1, a tumor suppressor and adaptor protein, is recurrently mutated in lung cancer.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links