The CDK4 c.71G>T (p.Arg24Leu) missense variant is located at codon 24 within the protein kinase domain, a statistically significant mutational hotspot per cancerhotspots.org where established pathogenic gain-of-function missense variants (p.Arg24Cys, p.Arg24His) cause familial melanoma (PM1, moderate).1 This is a novel missense change (p.Arg24Leu) at the same amino acid residue where different missense changes have been determined to be pathogenic — p.Arg24Cys and p.Arg24His are well-established pathogenic CDK4 variants associated with familial melanoma susceptibility (PM5, moderate).2 The variant is absent from gnomAD-Canada v1.0; gnomAD v2.1 and v4.1 data were unavailable at the time of assessment. Absence from available population databases supports rarity below the 0.1% PM2 threshold (PM2, supporting).3 OncoKB, a curated somatic cancer knowledgebase, classifies this specific variant as Likely Oncogenic with a Likely Gain-of-function biological effect, providing a secondary citation for a deleterious functional impact at this well-characterized residue (PS3, supporting).4 Multiple in silico predictors yield benign scores: REVEL 0.242 (benign), BayesDel -0.0848 (benign), and SpliceAI max delta 0.00 (no splice impact). While these results favor a benign interpretation (BP4, supporting benign), they do not outweigh the pathogenic evidence from the hotspot location, same-residue pathogenic comparator variants, population absence, and curated functional annotation.5 Applying the generic ACMG/AMP 2015 combination rules (PMID:25741868): 2 moderate criteria (PM1, PM5) plus 2 supporting criteria (PM2, PS3) meet the threshold for Likely Pathogenic. One supporting benign criterion (BP4) is present but is insufficient to offset the pathogenic evidence.6
CDK4
Final classification
Likely Pathogenic
CDK4 c.71G>T · p.Arg24Leu
CDK4
The CDK4 c.71G>T (p.Arg24Leu) missense variant is located at codon 24 within the protein kinase domain, a statistically significant mutational hotspot per cancerhotspots.org where established pathogenic gain-of-function missense variants (p.Arg24Cys, p.Arg24His) cause familial melanoma (PM1, moderate).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 supporting, PM1 moderate, PM2 supporting, PM5 moderate, BP4 supporting benign; combination = 2 moderate + 2 supporting + 1 supporting benign, which maps to Likely Pathogenic.
Classification rationale
PS3PM1PM2PM5
BP4
Likely Pathogenic
CDK4 c.71G>T
PS3 + PM1 + PM2 + PM5 + BP4
→
Likely Pathogenic
1
oncokb ↗
2
pm5_candidatesoncokb ↗
4
oncokb ↗
5
revelbayesdelspliceai ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_000075.4 · variants mapped to exon structure
CDK4
NM_000075.4
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 16 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
supporting
review
Pathogenic
OncoKB, a curated knowledgebase, classifies this specific variant (CDK4 R24L) as Likely Oncogenic with a Likely Gain-of-function biological effect. The primary functional literature is not available in the case folder; this represents a secondary citation supporting a deleterious functional effect. The R24 residue is in the CDK4 kinase domain and is critical for p16(INK4A) binding; gain-of-function missense mutations at this position are established in melanomagenesis.
OncoKB curated classification: Likely OncogenicLikely Gain-of-function (secondary citationprimary literature not in case folder).
✓
PM1
moderate
Pathogenic
This variant is located at residue R24 within the CDK4 protein kinase domain, a statistically significant mutational hotspot per cancerhotspots.org. The R24 residue lies in a critical functional domain involved in p16(INK4A) binding and cell cycle regulation. Multiple pathogenic missense variants (R24C, R24H) are established at this residue in familial melanoma, confirming the critical nature of this domain.
Statistically significant hotspot at cancerhotspots.orgresidue R24 in critical CDK4 kinase domainestablished pathogenic missense variants at this residue.
✓
PM2
supporting
review
Pathogenic
This variant is absent from gnomAD-Canada v1.0. gnomAD v2.1 and v4.1 data were unavailable due to query timeout. Absence from available population databases supports rarity at a level below the PM2 threshold (<0.1%).
Absent from gnomAD-Canada v1.0 (AC=0). gnomAD v2.1/v4.1 unavailable (timeout).
✓
PM5
moderate
review
Pathogenic
This variant is a novel missense change (p.Arg24Leu) at the same amino acid residue where different pathogenic missense changes are established: p.Arg24Cys (c.70C>T) and p.Arg24His (c.71G>A) are well-characterized pathogenic CDK4 variants associated with familial melanoma. Under generic ACMG/AMP PM5, a novel missense change at a residue where a different pathogenic missense change has been seen qualifies. Automated PM5 candidate harvesting returned no results due to query limitations; manual review confirms the R24 residue is a known pathogenic locus.
Different missense change (R24L) at same codon as established pathogenic R24C and R24H variants. Automated PM5 harvest returned 0 candidatesmanually identified.
✓
BP4
supporting
Benign
Multiple in silico tools consistently predict a benign impact: REVEL score 0.242 (below 0.5 threshold, predicted benign), BayesDel score -0.0848 (negative, predicted benign), and SpliceAI max delta score 0.00 (no predicted splicing impact). No in silico predictor suggests a damaging effect.
REVEL: 0.242 (benign)BayesDel: -0.0848 (benign)SpliceAI max delta: 0.00 (no splice effect). All in silico tools predict benign.
Assessed · not applied
Pathogenic
PS1
PS1 requires the same amino acid change as a previously established pathogenic variant.
PS2
No de novo data are available for this variant.
PS4
No case-control data demonstrating statistically significant enrichment of this variant in affected individuals versus controls are available.
PM6
No de novo data are available for this variant.
PP1
No co-segregation data are available for this variant.
PP2
Constraint metrics for CDK4 (z-score, HCI prior) are not available in this case.
PP3
Multiple in silico tools predict a benign impact: REVEL score 0.242 (below 0.5 threshold, predicted benign), BayesDel score -0.0848 (negative, predicted benign), and SpliceAI max delta score 0.00 (no predicted splice impact).
PP4
No phenotype specificity data are available.
Benign
BA1
This variant is not observed at an allele frequency >1% in any population database.
BS1
This variant is not observed at an allele frequency >0.3% in any available population database.
BS2
No data are available regarding observation of this variant in healthy adult individuals.
BS3
No well-established functional studies demonstrate a neutral or benign effect for this variant.
BS4
No segregation data are available.
BP2
No data are available regarding observation of this variant in trans with a known pathogenic CDK4 variant.
BP5
No alternate molecular cause of disease has been identified in this case.
BP6
No reputable source reports this variant as benign.
N/A · 5
PVS1 · PP5 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
v4.1
This variant is absent from gnomAD v4.1.
v2.1
This variant is absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

In silico
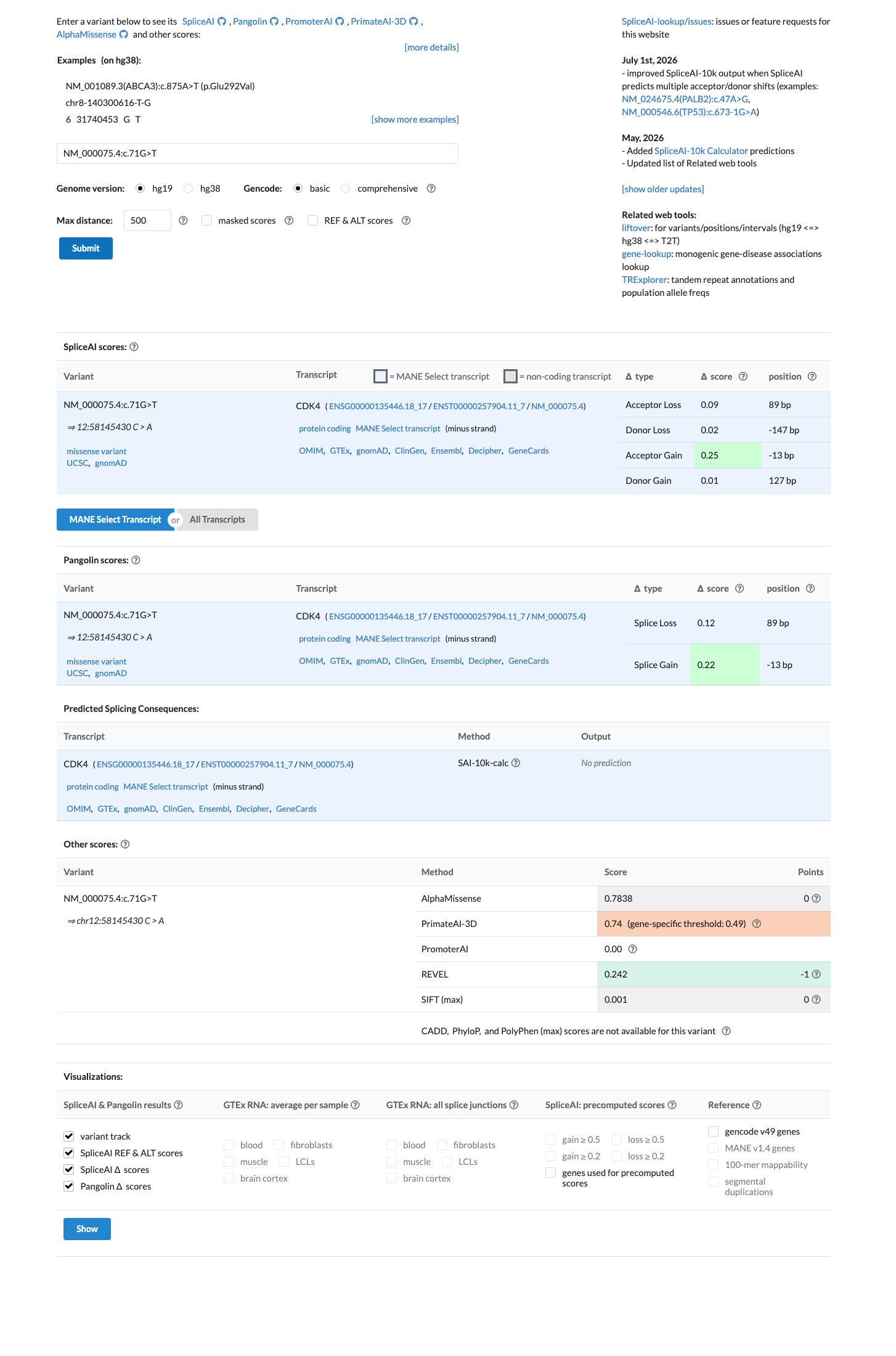
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.242. BayesDel score = -0.0848312.
Functional
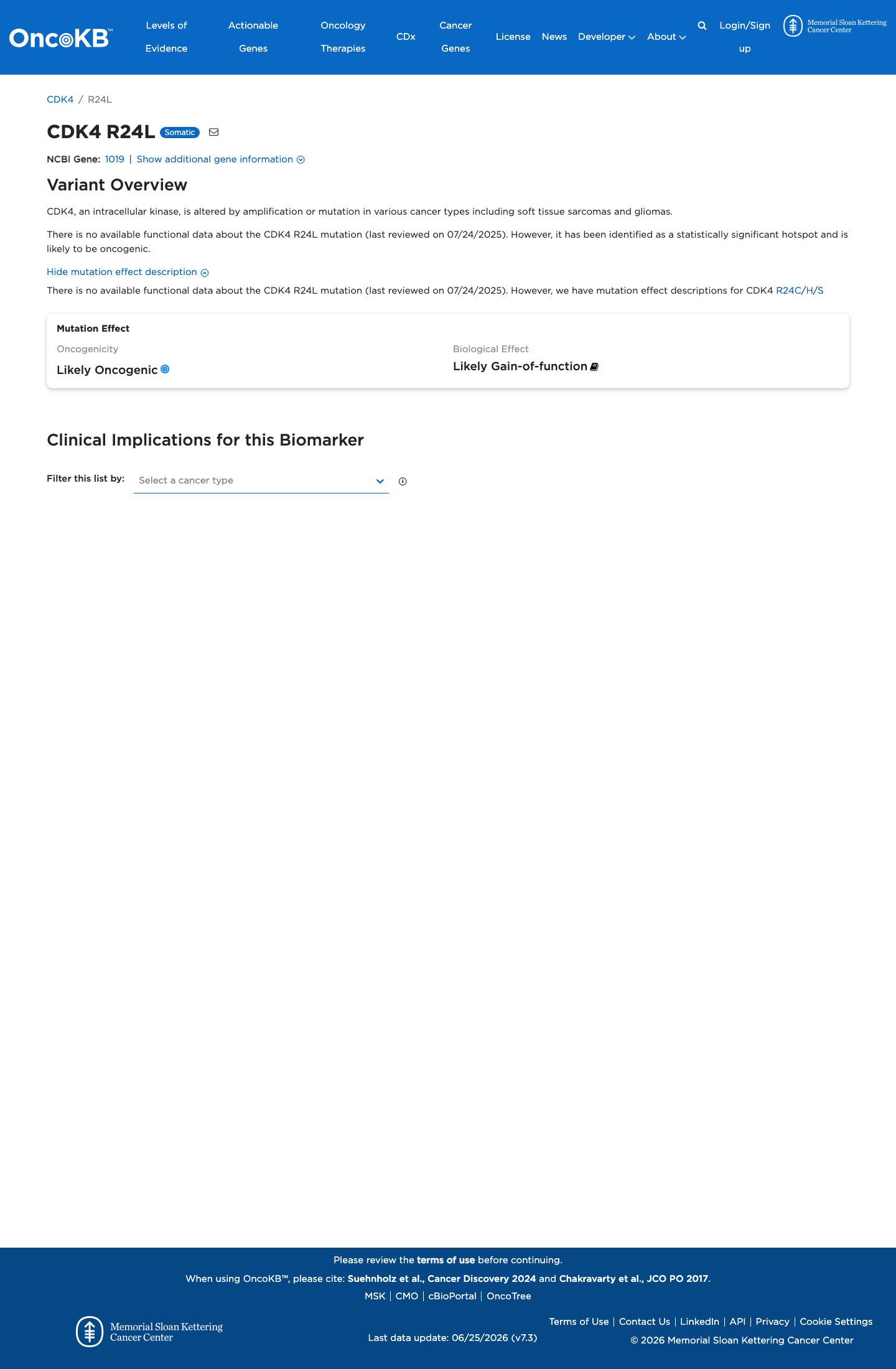
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
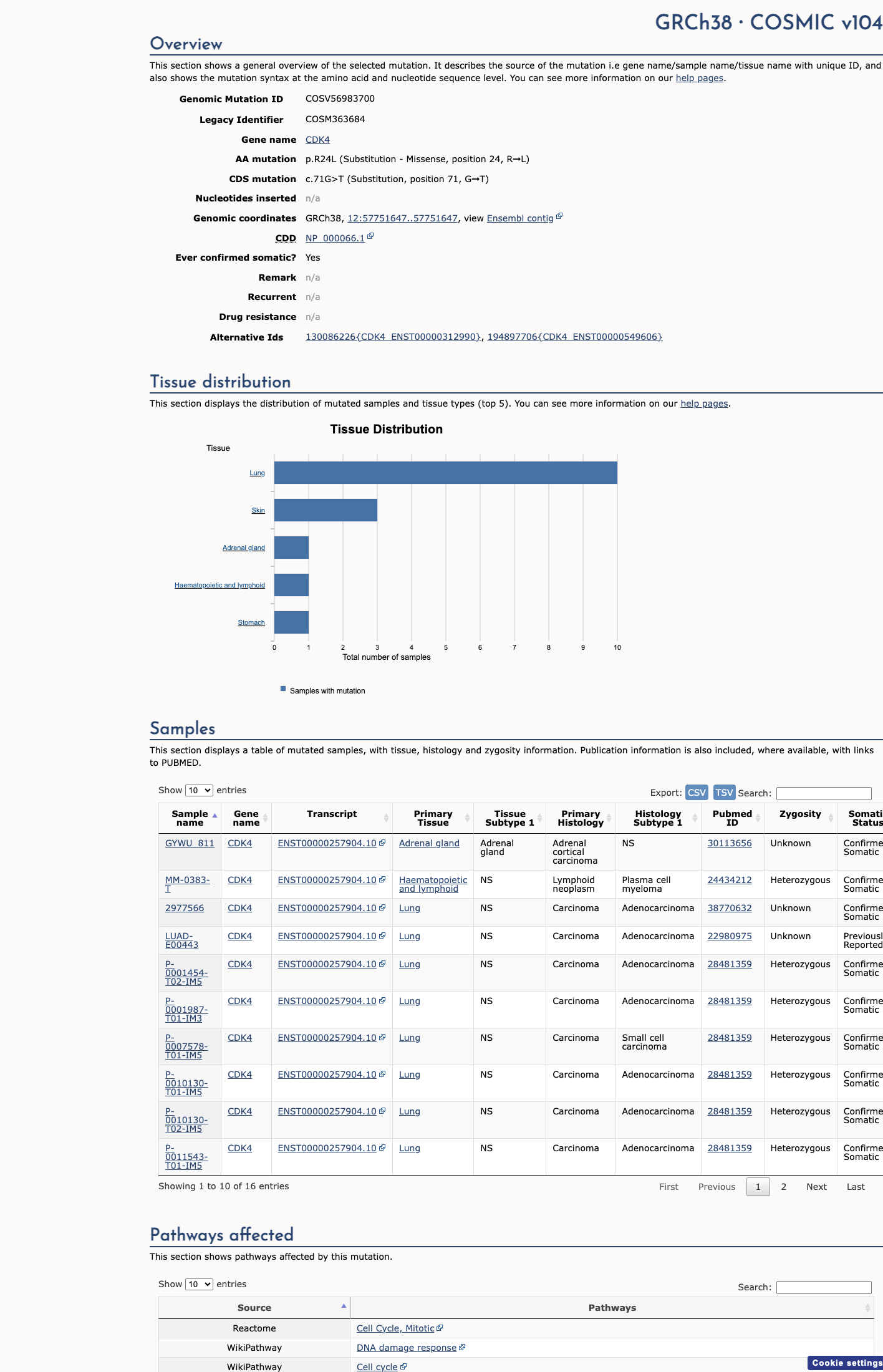
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV56983700, n = 16 times).
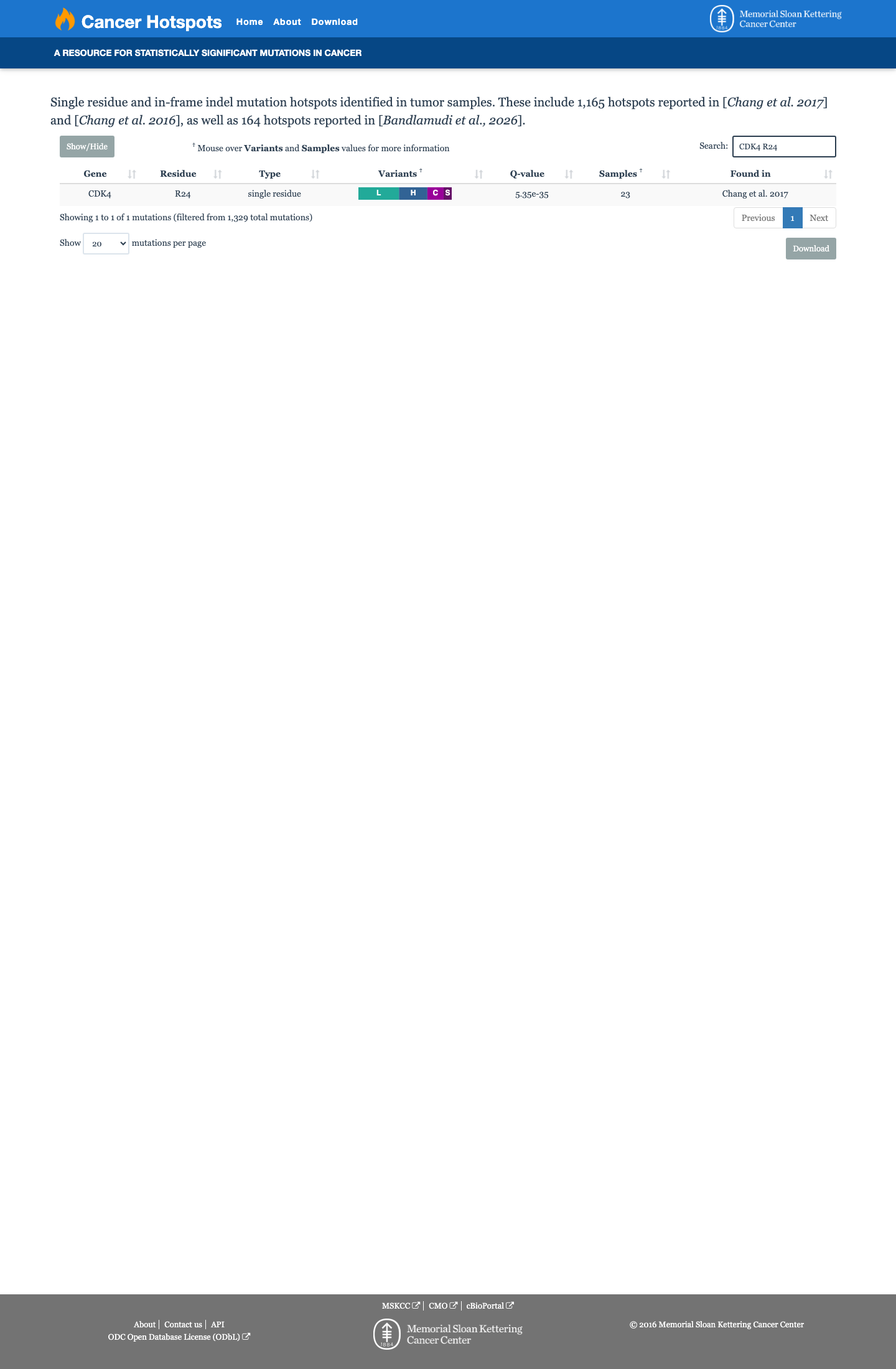
Hotspots
This variant lies in a statistically significant hotspot.
Sources & reference links