NM_000249.4:c.307-1G>A is a canonical splice acceptor variant (IVS3-1G>A) in MLH1, predicted to cause exon 4 skipping, frameshift, and nonsense-mediated decay.1 SpliceAI predicts a severe splicing defect with a max delta score of 0.99 (acceptor loss), consistent with disruption of the canonical splice acceptor.2 The variant is absent from gnomAD v4.1, v2.1, and gnomAD-Canada, indicating it is extremely rare in the general population.3 Under the InSiGHT VCEP v2.0.0 for MLH1, the variant qualifies for PVS1_Very_Strong (IVS±1 disrupting reading frame with predicted NMD) and PM2_Supporting (absent from gnomAD v4 with AF <0.00002).4 The variant has been reported in ClinVar as Likely pathogenic by two clinical laboratories (ClinVar Variation ID: 1727263) and has been observed once in somatic cancers (COSMIC: COSV99212388).5 No functional studies, co-segregation data, de novo observations, or tumor phenotype data are available for this specific variant in the reviewed literature.
MLH1
Final classification
VUS
MLH1 c.307-1G>A · p.?
MLH1
NM_000249.4:c.307-1G>A is a canonical splice acceptor variant (IVS3-1G>A) in MLH1, predicted to cause exon 4 skipping, frameshift, and nonsense-mediated decay.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MLH1 Version 2.0.0 v2.0.0 criteria-combination framework was evaluated deterministically with applied criteria: PVS1 very strong, PM2 supporting; no rule matched the adjudicated criteria.
Classification rationale
PVS1PM2
VUS
MLH1 c.307-1G>A
PVS1 + PM2
→
VUS
Gene diagram
· NM_000249.4 · variants mapped to exon structure
MLH1
NM_000249.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 12 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_000249.4:c.307-1G>A disrupts the canonical splice acceptor of intron 3 (IVS3-1G>A). Under the InSiGHT VCEP v2.0.0, variants at IVS±1 or IVS±2 where exon skipping or cryptic splice site use disrupts the reading frame and is predicted to undergo NMD are assigned PVS1_Very_Strong. Skipping of exon 4 (91 bp) would cause a frameshift and premature termination codon predicted to trigger nonsense-mediated decay. SpliceAI supports a severe splicing defect with a max delta score of 0.99 (acceptor loss). PP3 is not applied per VCEP guidance for IVS±1/2 variants receiving PVS1.
Canonical splice acceptor variant at IVS3-1 (c.307-1G>A)SpliceAI delta score 0.99 predicts acceptor lossExon 4 skipping (91 bp) disrupts reading frame
✓
PM2
supporting
Pathogenic
The variant is absent from gnomAD v4.1 (0 alleles in the population database). Under the InSiGHT VCEP v2.0.0, an allele frequency <0.00002 (<1 in 50,000 alleles) in gnomAD v4 qualifies for PM2_Supporting. The variant is also absent from gnomAD v2.1 and gnomAD-Canada.
Absent from gnomAD v4.1 (AF = 0.0meets <0.00002 threshold)Absent from gnomAD v2.1
Assessed · not applied
Pathogenic
PS2
No de novo observations have been reported for this variant.
PS3
No variant-specific functional data were identified.
PP1
No co-segregation data have been reported for this variant.
PP4
No tumor MSI/IHC phenotype data have been reported for patients carrying this variant.
Benign
BA1
BA1 requires gnomAD v4 Grpmax filtering allele frequency ≥0.001 (0.1%).
BS1
BS1 requires gnomAD v4 allele frequency ≥0.0001 and <0.001 (0.01-0.1%).
BS2
No evidence of co-occurrence in trans with a known pathogenic MLH1 variant in a patient without CMMRD features.
BS3
No functional studies demonstrating a benign effect for this variant have been identified.
BS4
No lack-of-segregation data have been reported.
BP4
BP4_Supporting requires HCI prior probability <0.11 for missense variants, or SpliceAI delta score ≤0.1 for intronic/synonymous variants.
BP5
No tumor data showing MSS status or normal MMR protein expression have been reported for patients carrying this variant.
BP7
BP7 applies to intronic variants at or beyond -21/+7 positions.
N/A · 11
PS1 · PS4 · PM1 · PM5 · PM6 · PP2 · PP3 · PP5 · BP1 · BP2 · BP6
Research & evidence
Population frequency
gnomAD v4.1

gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
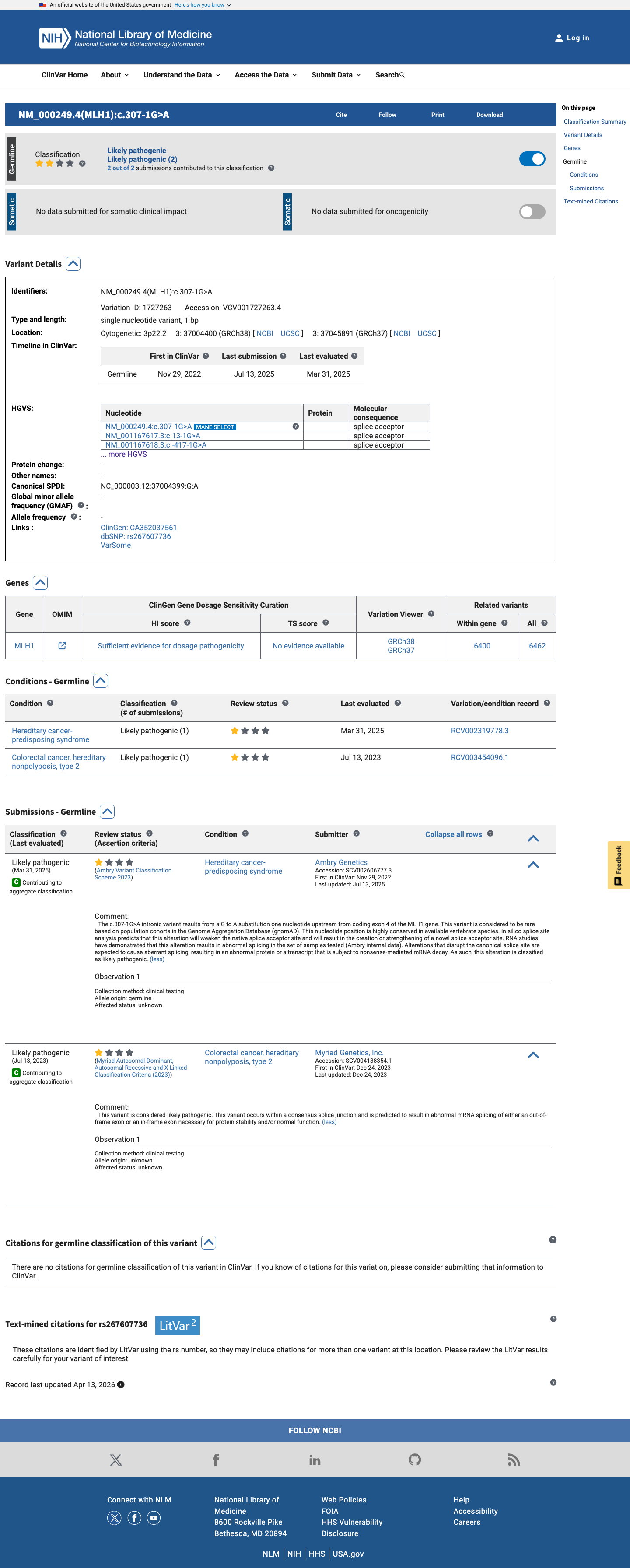
ClinVar
This variant has been reported in ClinVar as Likely pathogenic (2 clinical laboratories). (ClinVarID = 1727263)
In silico
SpliceAI predicts possible splice impact for this variant (max delta score = 0.99). BayesDel score = 0.66.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
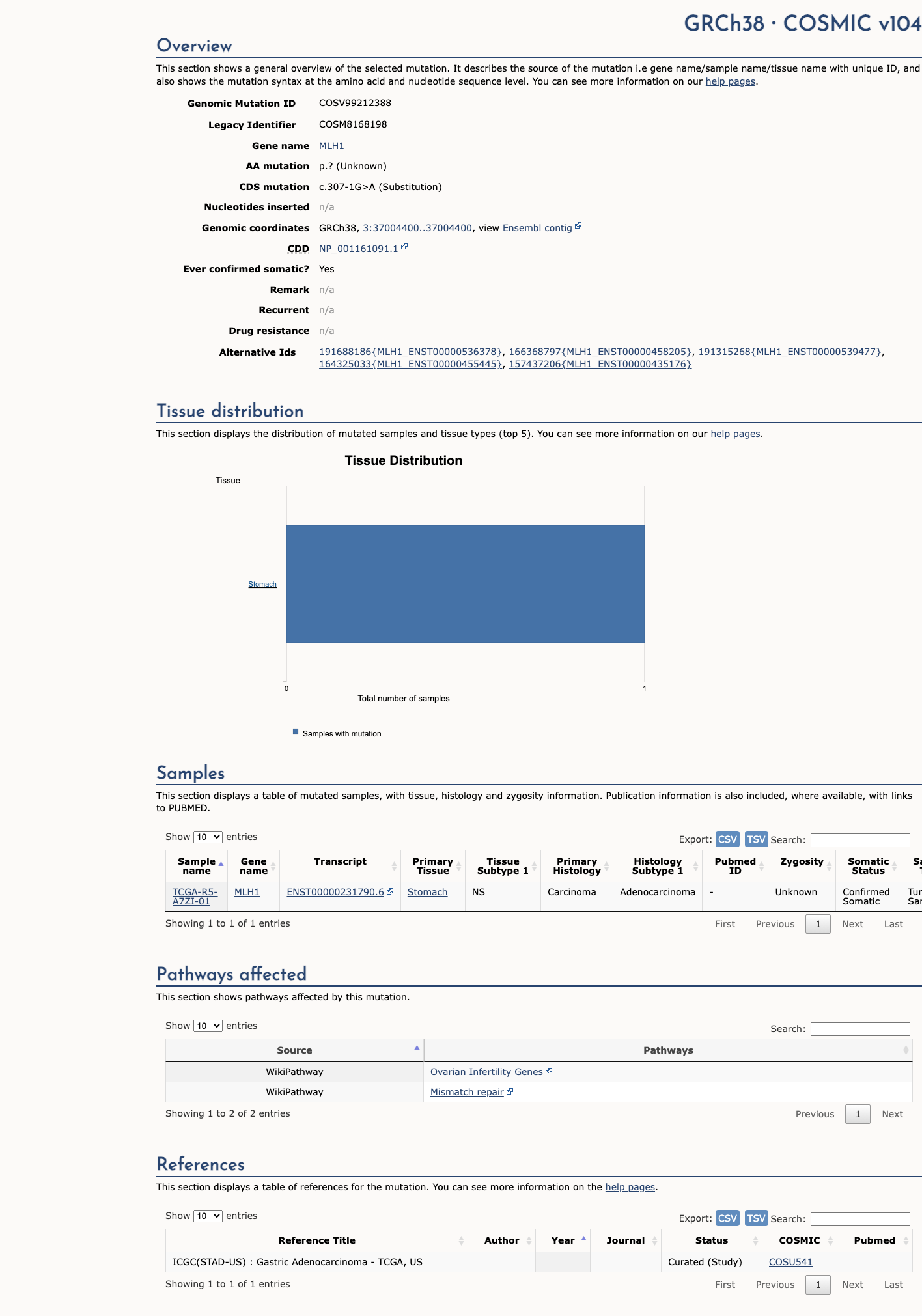
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV99212388, n = 1 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
22167527 ↗
Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline.
CLINVAR
23535968 ↗
Informing family members of individuals with Lynch syndrome: a guideline for clinical geneticists.
CLINVAR
23788249 ↗
ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.
CLINVAR
25356965 ↗
ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing.
CLINVAR
25452455 ↗
Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines.
CLINVAR