NM_018062.3:c.1A>G (p.Met1Val) is an initiation codon variant in FANCL, where loss-of-function is an established mechanism for autosomal recessive Fanconi anemia. The variant abolishes the initiator methionine with no alternative start codon in exon 1, qualifying for PVS1 at moderate strength per ClinGen SVI recommendations (PMC6185798).1 The variant is absent from gnomAD v2.1 (0/250,782 alleles) and present at extremely low frequency in gnomAD v4.1 (AF=0.00037%, 6/1,614,100 alleles, no homozygotes), meeting PM2 at supporting strength.2 Two clinical laboratories have independently classified this variant as Likely pathogenic and Pathogenic in ClinVar (Variation ID 1691887, 2-star review status), satisfying PP5 at supporting strength. However, full-text review of the PMIDs cited in these ClinVar submissions did not confirm variant-specific evidence.3 No variant-specific functional studies (PS3), de novo observations (PS2/PM6), segregation data (PP1), or case-control data (PS4) were identified for this variant in the reviewed literature. The ACMG/AMP evidence tally of 1 moderate (PVS1) and 2 supporting (PM2, PP5) criteria does not reach the Likely Pathogenic threshold (requires 1 moderate + 4 supporting or 2 moderate + 2 supporting). This variant is classified as a Variant of Uncertain Significance (VUS) by formal ACMG/AMP 2015 combination rules, bordering on Likely Pathogenic given the initiation codon loss mechanism and concordant clinical laboratory classifications.4
FANCL
Final classification
VUS
FANCL c.1A>G · p.Met1?
FANCL
NM_018062.3:c.1A>G (p.Met1Val) is an initiation codon variant in FANCL, where loss-of-function is an established mechanism for autosomal recessive Fanconi anemia. The variant abolishes the initiator methionine with no alternative start codon in exon 1, qualifying for PVS1 at moderate strength per ClinGen SVI recommendations (PMC6185798).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 moderate, PM2 supporting, PP5 supporting; combination = 1 moderate + 2 supporting, which maps to VUS.
Classification rationale
PVS1PM2PP5
VUS
FANCL c.1A>G
PVS1 + PM2 + PP5
→
VUS
1
pvs1_generic_framework ↗pvs1_gene_contextPMID:25754594 ↗
Gene diagram
· NM_018062.3 · variants mapped to exon structure
FANCL
NM_018062.3
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 18 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
moderate
review
Pathogenic
c.1A>G is an initiation codon variant (ATG>GTG, p.Met1Val) in FANCL, a gene where loss-of-function is an established disease mechanism for Fanconi anemia. No alternative in-frame ATG start codon is present in exon 1 (residues 1-32: MAVTEASLLRQCPLLLPQNRSKTVYEGFISAQ). Per ClinGen SVI PVS1 recommendations (PMC6185798), initiation codon variants without an alternative start codon in the same exon qualify for PVS1 at moderate strength. The pipeline-generated pvs1_variant_assessment incorrectly assigned this variant to the 'other' bucket; this adjudication overrides that assignment because c.1A>G is unequivocally a start codon loss, which is an ACMG-recognized null variant type.
FANCL loss-of-function is an established disease mechanism for Fanconi anemiasupported by truncating mutations reported in FA patients (PMID:25754594) and zebrafish knockout models (PMID:30540754)c.1A>G alters the initiator ATG to GTG
✓
PM2
supporting
Pathogenic
NM_018062.3:c.1A>G is absent from gnomAD v2.1 (0/250,782 alleles) and present at extremely low frequency in gnomAD v4.1 (AF=0.00037%, 6/1,614,100 alleles, no homozygotes). The maximum observed subpopulation frequency is in European (non-Finnish) at 0.00051% (6/1,180,000). The grpmax filtering allele frequency (FAF) is 1.83e-06. All frequencies are well below the PM2 threshold of <0.1%. The variant is also absent from gnomAD-Canada v1.0. Assigned supporting rather than moderate strength due to trace presence (6 alleles) in gnomAD v4.1.
gnomAD v2.1: 0/250782 alleles (0%)gnomAD v4.1: 6/1
✓
PP5
supporting
review
Pathogenic
Two clinical diagnostic laboratories have independently classified NM_018062.3:c.1A>G as likely pathogenic or pathogenic in ClinVar (Variation ID: 1691887; VCV001691887). Labcorp Genetics/Invitae (SCV005703968) classifies as Pathogenic with criteria provided (citing PM2, PS3). Fulgent Genetics (SCV005658736) classifies as Likely pathogenic (clinical testing). The ClinVar aggregate classification is Pathogenic/Likely pathogenic with review status 'criteria provided, multiple submitters, no conflicts' (2 stars). However, these classifications are from single submitters without expert panel review, and the evidence underlying the cited criteria is not independently verifiable from the case materials. Full-text review of the cited PMIDs did not confirm variant-specific evidence. Assigned supporting strength per ACMG/AMP PP5 guidance for reputable source reporting without independent verification.
ClinVar Variation ID 1691887: Pathogenic/Likely pathogenic2 starscriteria provided by multiple submitters with no conflicts
Assessed · not applied
Pathogenic
PS2
No de novo observation of NM_018062.3:c.1A>G has been reported in the literature.
PS3
No variant-specific functional studies were identified.
PS4
No case-control or cohort data comparing the prevalence of NM_018062.3:c.1A>G in affected individuals versus the general population has been published.
PM1
The variant affects the initiator methionine at the very N-terminus of FANCL.
PM6
No de novo confirmation of NM_018062.3:c.1A>G has been reported in the literature.
PP1
No segregation data are available for NM_018062.3:c.1A>G.
PP2
FANCL disease mechanism is loss-of-function, not missense-predominant.
PP3
REVEL score 0.227 falls below the typical pathogenic threshold (>0.5).
PP4
While Fanconi anemia is a highly specific clinical syndrome, its genetic etiology involves at least 22 genes (FANCA through FANCW).
Benign
BA1
The maximum allele frequency of NM_018062.3:c.1A>G in gnomAD is 0.00051% (European non-Finnish, v4.1), far below the BA1 threshold of >1%.
BS1
The maximum allele frequency of NM_018062.3:c.1A>G in gnomAD is 0.00051% (European non-Finnish, v4.1), far below the BS1 threshold of >0.3%.
BS2
No observation of NM_018062.3:c.1A>G in a healthy adult individual has been documented outside of population databases.
BS3
No well-established in vitro or in vivo functional studies demonstrate that NM_018062.3:c.1A>G has no deleterious effect on protein function or splicing.
BS4
No lack of segregation data has been reported for NM_018062.3:c.1A>G.
BP2
No evidence demonstrates that NM_018062.3:c.1A>G has been observed in trans with a known pathogenic FANCL variant in a healthy individual.
BP4
While BayesDel (-0.184843) and REVEL (0.227) scores are low and SpliceAI predicts no splicing impact (delta 0.00), these in silico tools are designed and calibrated for standard missense variants.
BP5
No observation of NM_018062.3:c.1A>G in a case where an alternative molecular basis for disease was identified has been reported.
BP6
No reputable source classifies NM_018062.3:c.1A>G as benign or likely benign.
N/A · 4
PS1 · PM5 · BP1 · BP7
Research & evidence
Population frequency
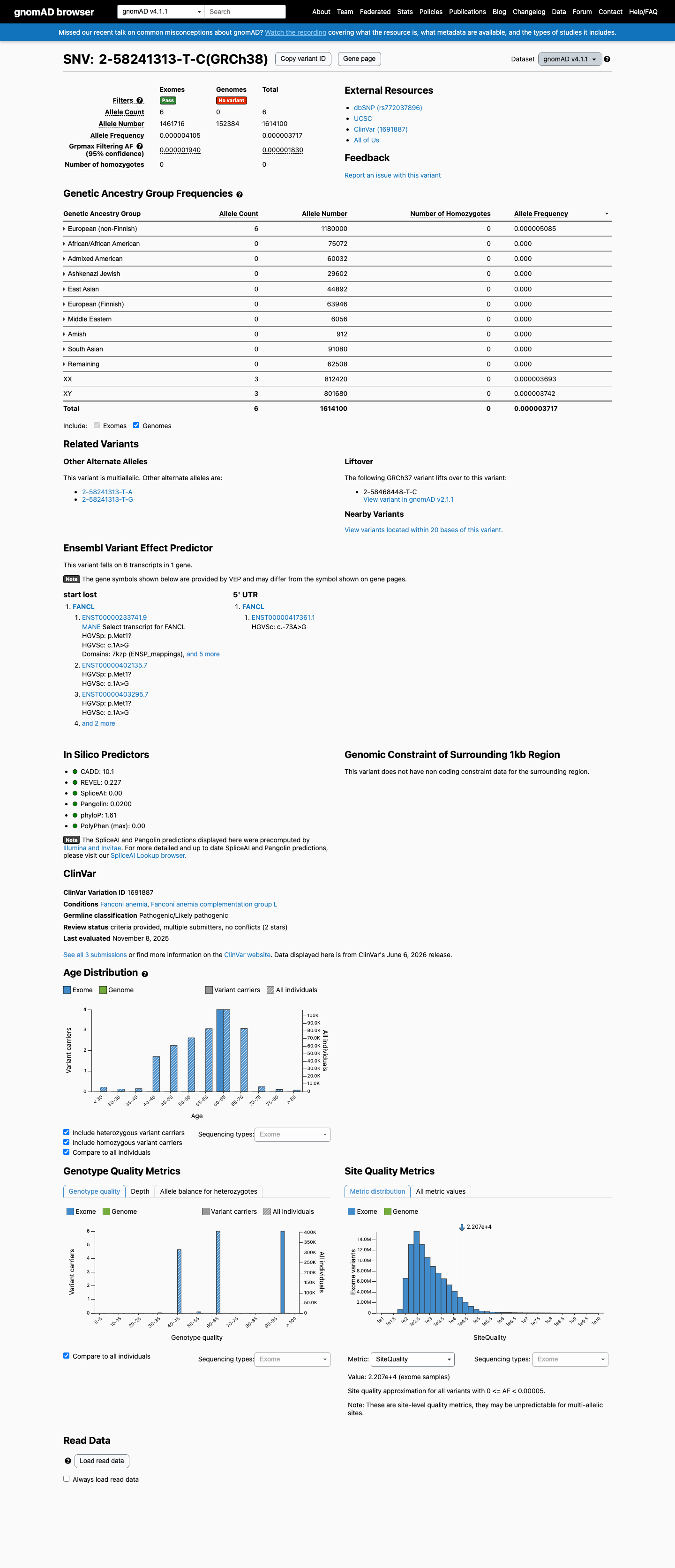
gnomAD v4.1
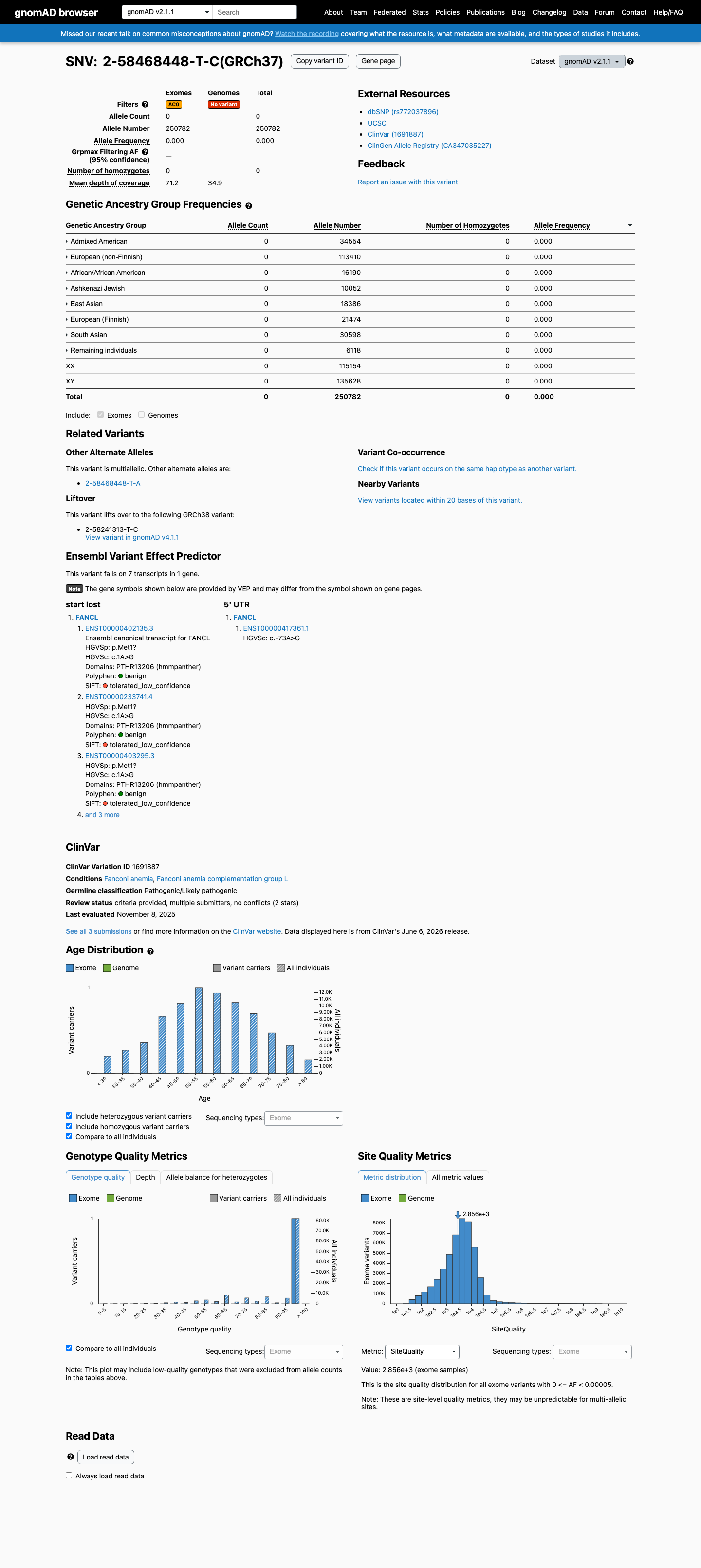
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 3.71724e-06; MAF= 0.00037%, 6/1614100 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 5.08475e-06; MAF= 0.00051%, 6/1180000 alleles, homozygotes = 0); grpmax FAF= 1.83e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/250782 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/16190 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00037%
· 6 / 1,614,100
0 hom · FAF 0.00018%
0 hom · FAF 0.00018%
European (non-Finnish) 6 / 1,180,000 |
0.00051% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / 250,782
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
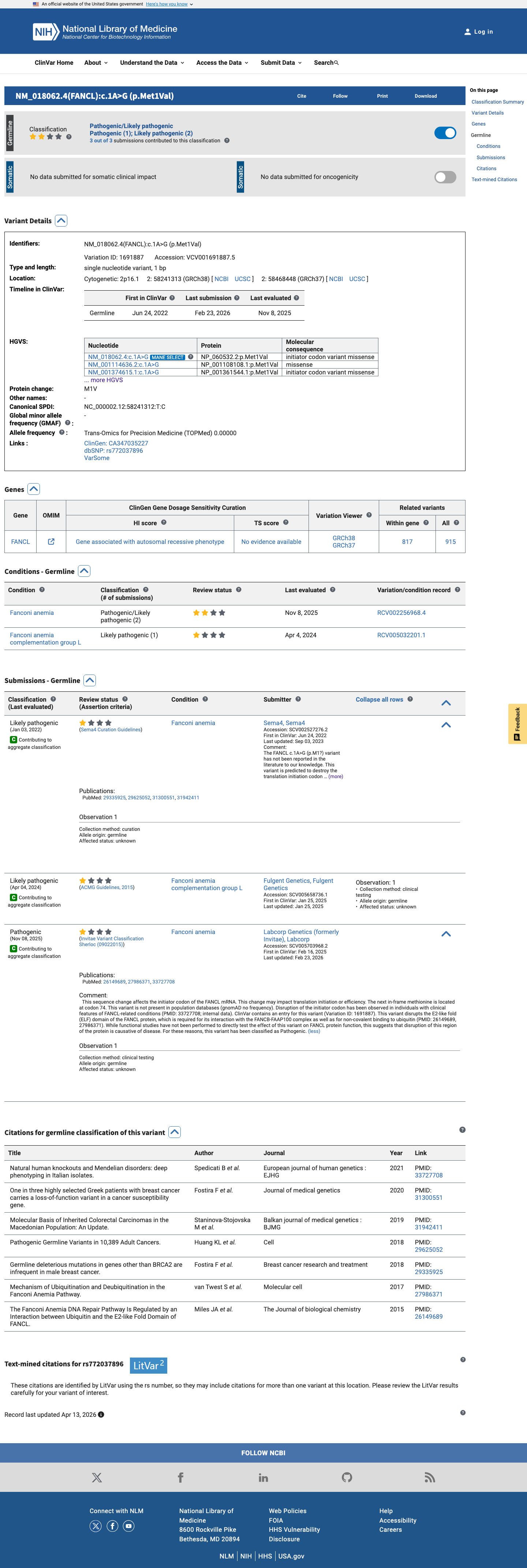
ClinVar
This variant has been reported in ClinVar as Likely pathogenic (1 clinical laboratory) and as Pathogenic (1 clinical laboratory). (ClinVarID = 1691887)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.227. BayesDel score = -0.184843.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
3papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 5 further PMIDs triaged but not cited — see Sources & References.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Searched
c.1A>GFANCLMet1
Found
ACMG/AMP standards and guidelines for the interpretation of sequence variants. This is a methodological guideline document and does not discuss any specific FANCL variants.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PVS1 supports · met
Why
Framework document; cited for PVS1 criterion methodology (initiation codon = null variant type) but does not contain variant-specific evidence.
Location Full text reviewed; no variant-specific content · full text
Loss-of-Function FANCL Mutations Associate with Severe Fanconi Anemia Overlapping the VACTERL Association.
Searched
c.1A>Gp.Met1ValM1Vstart codonc.1A
Found
Reports two novel FANCL truncating mutations (c.268del, p.Leu90Phefs*6 and c.430del, p.Ser144Leufs*6) in three patients with severe Fanconi anemia overlapping VACTERL association. Both mutations cause NMD and loss of FANCD2 monoubiquitination; functional complementation with wild-type FANCL rescued the phenotype. NM_018062.3:c.1A>G was not among the mutations identified.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PVS1 supports · met
Why
Establishes FANCL loss-of-function as disease mechanism in Fanconi anemia; cited for PVS1 gene-level LOF evidence. Variant-specific evidence not present.
a single nucleotide deletion (NM_018062.3:c.268del; p.Leu90Phefs*6) in exon 4 of FANCL
Location Full text reviewed; two other FANCL variants (c.268del, c.430del) reported but not c.1A>G · full text
Multiplexed CRISPR/Cas9-mediated knockout of 19 Fanconi anemia pathway genes in zebrafish revealed their roles in growth, sexual development and fertility.
Searched
c.1A>GM1VMet1Valstart codonc.1A
Found
Used multiplexed CRISPR/Cas9 to generate loss-of-function knockout mutants for 17 FA pathway genes in zebrafish (including fancl). Generated two independent indel mutant lines for fancl predicted to cause premature truncations. This is a systematic gene-level knockout study; no specific human FANCL missense or initiation codon variants were tested.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PVS1 supports · met
Why
Establishes FANCL loss-of-function as disease mechanism through systematic zebrafish knockout; cited for PVS1 gene-level LOF support. No variant-specific data.
Location Full text reviewed; fancl knockout zebrafish generated but no human c.1A>G variant tested · full text
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
20661450 ↗
Sex reversal in zebrafish fancl mutants is caused by Tp53-mediated germ cell apoptosis.
ONCOKB
26149689 ↗
The Fanconi Anemia DNA Repair Pathway Is Regulated by an Interaction between Ubiquitin and the E2-like Fold Domain of FANCL.
CLINVAR
29335925 ↗
Germline deleterious mutations in genes other than BRCA2 are infrequent in male breast cancer.
CLINVAR