NM_002878.3:c.26G>C (p.Cys9Ser) is a missense variant in exon 1 of RAD51D. This variant is present in gnomAD at a low frequency: 0.0405% (113/278,930 alleles, 1 homozygote) in v2.1 and 0.0656% (1058/1,612,440 alleles, 1 homozygote) in v4.1, with a maximum population frequency of 0.083% in the European (non-Finnish) population (v4.1).1 The variant has been reported in ClinVar (ID 127886) as Uncertain significance by 10 clinical laboratories and Likely benign by 9 clinical laboratories; no expert panel has reviewed this variant.2 Multiple lines of computational evidence predict no significant impact on the gene product: REVEL score 0.218, BayesDel score 0.087, and SpliceAI maximum delta score 0.00.3 PM2 (supporting) is applied because the variant is present at very low frequency in population databases (<0.1% AF in all populations), though weakened by the observation of homozygotes.4 BP4 (supporting benign) is applied because multiple computational prediction algorithms (REVEL, BayesDel, SpliceAI) support no deleterious effect.5 BP1 (supporting benign) is applied because RAD51D-associated disease is primarily caused by truncating loss-of-function variants rather than missense changes.6 BS2 (supporting benign) is applied because one homozygous individual was observed in gnomAD v2.1 and one in gnomAD v4.1, suggesting this variant can be tolerated in at least some individuals.7 BP6 (supporting benign) is applied because nine independent clinical laboratories have classified this variant as Likely benign with criteria provided.8 PVS1 is not applicable as this is a missense variant, not a null variant.9 PS3 is not met because no experimental functional studies have been performed for this variant.10 PP3 is not met because the aggregate computational evidence supports a benign rather than pathogenic interpretation.11 Using generic ACMG/AMP 2015 combination rules (PMID:25741868), the evidence profile includes: 1 supporting pathogenic criterion (PM2) and 4 supporting benign criteria (BP1, BP4, BS2, BP6). The net benign evidence outweighs the single pathogenic criterion, supporting a classification of Likely benign.12
RAD51D
Final classification
Likely Benign
RAD51D c.26G>C · p.Cys9Ser
RAD51D
NM_002878.3:c.26G>C (p.Cys9Ser) is a missense variant in exon 1 of RAD51D.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BS2 supporting benign, BP1 supporting benign, BP4 supporting benign, BP6 supporting benign; combination = 1 supporting + 4 supporting benign, which maps to Likely Benign.
Classification rationale
PM2
BS2BP1BP4BP6
Likely Benign
RAD51D c.26G>C
PM2 + BS2 + BP1 + BP4 + BP6
→
Likely Benign
3
revelbayesdelspliceai ↗
5
revelbayesdelspliceai ↗
9
pvs1_variant_assessment
10
oncokb ↗
11
revelbayesdelspliceai ↗
12
generic_acmg_combination_rules
Gene diagram
· NM_002878.3 · variants mapped to exon structure
RAD51D
NM_002878.3
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 17 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present in gnomAD at very low frequency: v2.1 AF = 0.0405% (113/278,930 alleles) and v4.1 AF = 0.0656% (1058/1,612,440 alleles), with grpmax FAF of 0.065% (v2.1) and 0.079% (v4.1), all below the 0.1% PM2 threshold for non-VCEP assessment. However, the presence of one homozygote in each database and over 100 observed alleles weakens the confidence; thus applied at supporting strength only.
gnomAD v2.1: AF = 0.0405%grpmax FAF = 0.065%.gnomAD v4.1: AF = 0.0656%
✓
BS2
supporting
Benign
One homozygous individual is observed in gnomAD v2.1 and one homozygous individual is observed in gnomAD v4.1. RAD51D-associated cancer predisposition is adult-onset with moderate penetrance, so homozygous observation does not definitively exclude pathogenicity, but it provides supporting evidence that the variant can be tolerated in at least some individuals. Applied at supporting benign strength.
gnomAD v2.1: 1 homozygote (exome).gnomAD v4.1: 1 homozygote.
✓
BP1
supporting
Benign
RAD51D is a gene in which the primary disease mechanism is loss-of-function through truncating variants (nonsense, frameshift, canonical splice). The ACMG/AMP 2015 guidelines (PMID:25741868) specify BP1 for missense variants in genes where primarily truncating variants cause disease. The RAD51D locus is well-characterized as an ovarian cancer susceptibility gene where the vast majority of reported pathogenic variants are protein-truncating.
RAD51D disease mechanism is predominantly loss-of-function via truncating variants.ACMG/AMP 2015 BP1 rule applies to missense variants in LOF-predominant genes.
✓
BP4
supporting
Benign
Multiple lines of computational evidence support a benign interpretation. REVEL score is 0.218 (benign-leaning, well below the 0.5 threshold). BayesDel score is 0.087 (low, consistent with benign). SpliceAI delta score is 0.00 (no predicted splicing impact). SIFT predicts tolerated (PMID:24130102). The aggregate in silico evidence indicates no significant impact on gene product.
REVEL = 0.218.BayesDel = 0.087.SpliceAI max delta = 0.00.
✓
BP6
supporting
Benign
Nine clinical laboratories in ClinVar have classified this variant as Likely benign (variation ID 127886). Multiple reputable clinical testing laboratories independently arrived at a likely benign classification with criteria provided, including Ambry Genetics, LabCorp, and Quest Diagnostics. This represents a substantial body of clinical laboratory consensus favoring a benign interpretation.
ClinVar ID 127886: 9 clinical laboratories report Likely benign.Multiple laboratories with criteria provided (Ambry Genetics SCV000185831LabCorp SCV000698094
Assessed · not applied
Pathogenic
PS1
No evidence was identified that a different nucleotide change at codon 9 resulting in the same amino acid substitution (Cys9Ser) has been previously reported as pathogenic.
PS2
No de novo data are available for this variant.
PS3
No experimental functional studies have been performed for NM_002878.3:c.26G>C (p.Cys9Ser).
PS4
No case-control study demonstrates statistically significant enrichment of this variant in affected individuals compared to controls.
PM1
Codon 9 (Cys9) lies in the N-terminal region of RAD51D, outside any established critical functional domain.
PM6
No de novo data are available for this variant.
PP1
No co-segregation data are available for this variant.
PP2
RAD51D-associated disease is primarily driven by loss-of-function (truncating) variants.
PP3
Multiple lines of computational evidence predict no damaging effect.
PP4
No proband phenotype or clinical history was provided for the index case.
PP5
No reputable source has classified this variant as pathogenic.
Benign
BA1
The maximum observed allele frequency in gnomAD is 0.083% (v4.1 NFE), well below the 1% BA1 threshold.
BS1
The maximum observed allele frequency in gnomAD is 0.083% (v4.1 NFE), below the 0.3% BS1 threshold for non-VCEP assessment.
BS3
No in vitro or in vivo functional studies have been performed to evaluate the effect of p.Cys9Ser on RAD51D protein function.
BS4
No co-segregation data are available to evaluate lack of segregation with disease in affected families.
BP2
No data are available regarding observation of this variant in trans with a known pathogenic variant in RAD51D.
BP5
No data are available regarding an alternate molecular basis for disease in a proband carrying this variant.
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency · supports benign

gnomAD v4.1
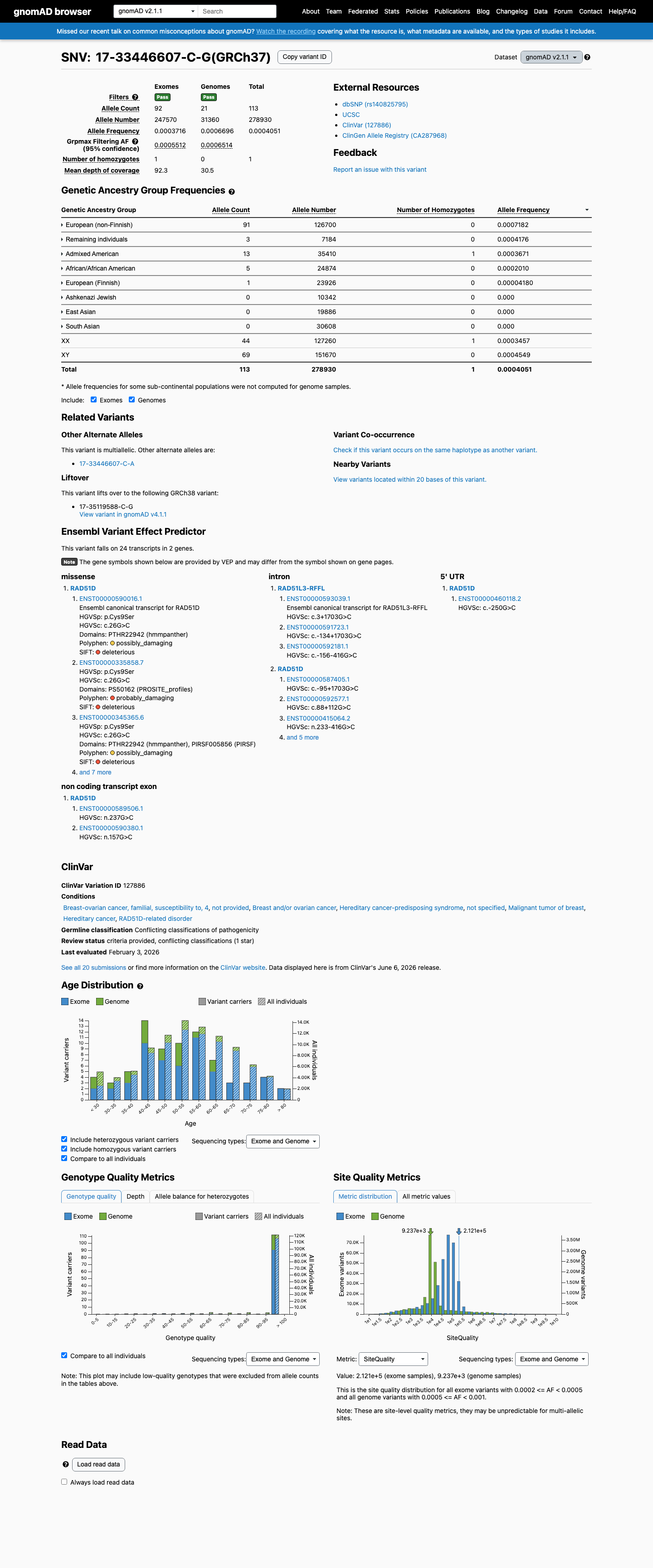
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000656148; MAF= 0.06561%, 1058/1612440 alleles, homozygotes = 1) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000830546; MAF= 0.08305%, 980/1179946 alleles, homozygotes = 0); grpmax FAF= 0.00078695.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00040512; MAF= 0.04051%, 113/278930 alleles, homozygotes = 1) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000718232; MAF= 0.07182%, 91/126700 alleles, homozygotes = 0); grpmax FAF= 0.00065141.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0011406844106463879, 21/18410 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.066%
· 1058 / 1,612,440
1 hom · FAF 0.079%
1 hom · FAF 0.079%
European (non-Finnish) 980 / 1,179,946 |
0.083% |
Remaining individuals 49 / 62,474 |
0.078% |
Admixed American 15 / 60,030 |
0.025% 1 hom |
African/African American 12 / 75,044 |
0.016% |
European (Finnish) 2 / 62,412 |
0.0032% |
+ 5 not observed (Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.041%
· 113 / 278,930
1 hom · FAF 0.065%
1 hom · FAF 0.065%
European (non-Finnish) 91 / 126,700 |
0.072% |
Remaining individuals 3 / 7,184 |
0.042% |
Admixed American 13 / 35,410 |
0.037% 1 hom |
African/African American 5 / 24,874 |
0.02% |
European (Finnish) 1 / 23,926 |
0.0042% |
+ 3 not observed (Ashkenazi Jewish, East Asian, South Asian)
gnomAD Canada 🇨🇦
0.11%
· 21 / 18,410
0 hom · FAF 0.042%
0 hom · FAF 0.042%
Latino/Admixed American 2 / 838 |
0.24% |
European (non-Finnish) 19 / 11,732 |
0.16% |
+ 7 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
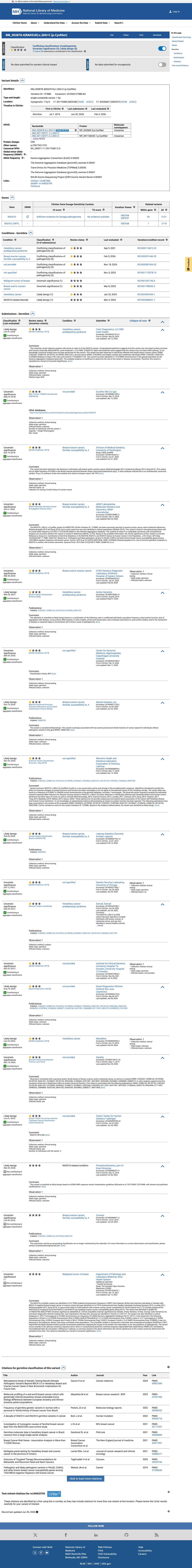
ClinVar
This variant has been reported in ClinVar as Uncertain significance (10 clinical laboratories) and as Likely benign (9 clinical laboratories). (ClinVarID = 127886)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.218. BayesDel score = 0.0874681.
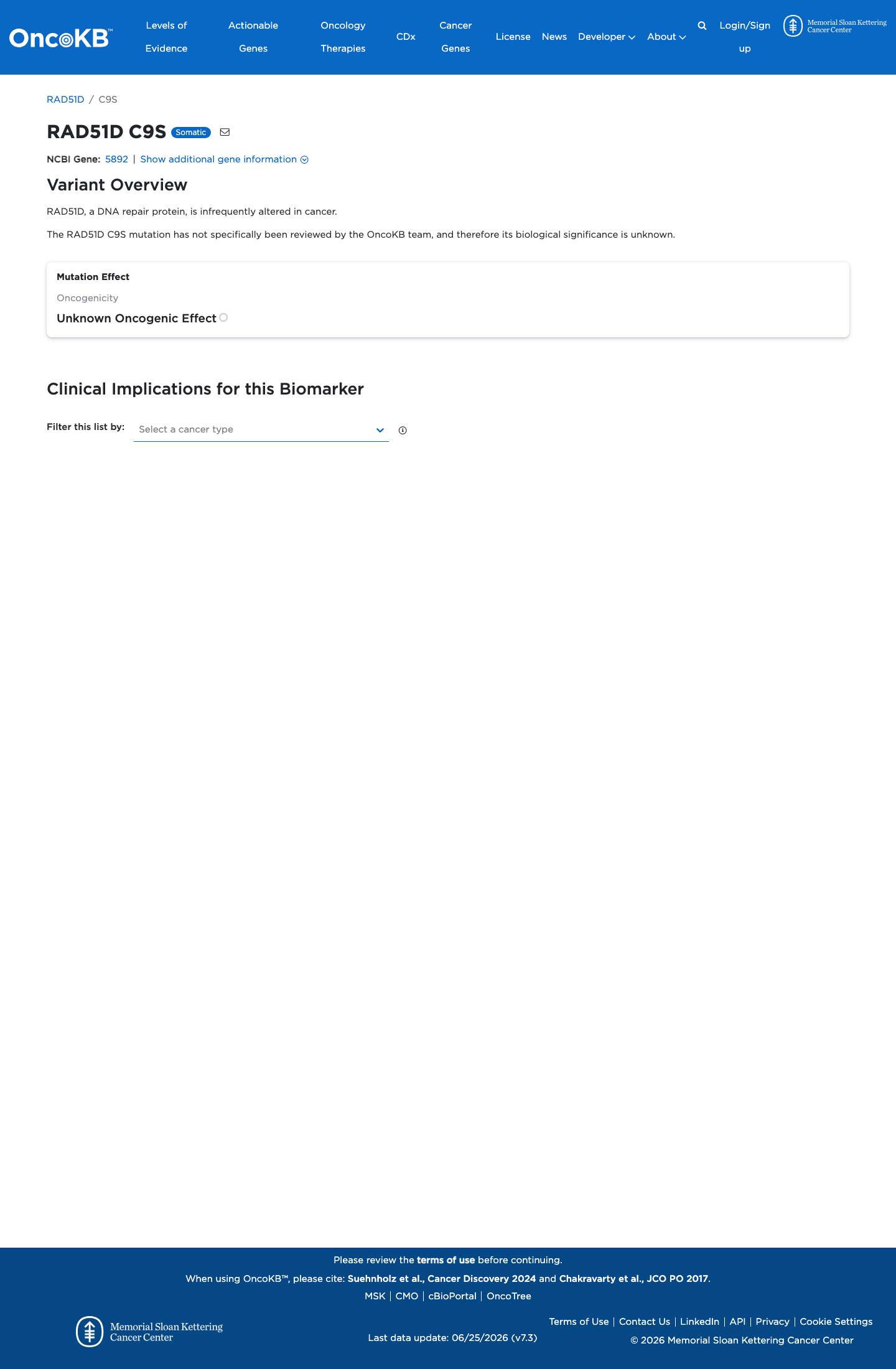
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. RAD51D, a DNA repair protein, is infrequently altered in cancer.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 7 further PMIDs triaged but not cited — see Sources & References.
About 1% of the breast and ovarian Spanish families testing negative for BRCA1 and BRCA2 are carriers of RAD51D pathogenic variants.
Searched
c.26G>Cp.Cys9SerCys9SerC9S26G>C
Found
Screened 842 Spanish BC/OC families for RAD51D mutations. c.26G>C (p.Cys9Ser, rs140825795) was identified as a nonsynonymous variant in 1/842 index cases (MAF 0.0006). In silico predictions: SIFT tolerated, PolyPhen possibly damaging, Condel deleterious, no splicing effect. Not considered pathogenic by the authors; listed separately from the three clearly pathogenic/likely pathogenic mutations identified.
Variant
✓ Names this variant — characterised directly
Applied to
→BP4 supports · met
Why
Variant identified as a nonsynonymous change of uncertain significance. In silico predictions were mixed (SIFT tolerated vs Condel deleterious); combined with REVEL and BayesDel, the aggregate supports BP4.
c.26G>C p.Cys9Ser rs140825795 C0 Tolerated Possibly damaging Deleterious No effect 0.0006 (842)
Location Table 1 (Nonsynonymous variants section) · full text
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26261251 ↗
Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population.
CLINVAR
33203166 ↗
Outcome of Targeted Therapy Recommendations for Metastatic and Recurrent Head and Neck Cancers.
CLINVAR