NM_001042749.1:c.1018-1G>A is a canonical splice acceptor variant in STAG2, where loss of function is a known disease mechanism. The variant is assigned PVS1 at very strong strength under ClinGen SVI PVS1 recommendations (PMC6185798), supported by SpliceAI delta 1.00 predicting abolition of the canonical 3' acceptor site.1 This variant is completely absent from gnomAD v2.1 and v4.1 (0/506,187 alleles across all populations), meeting PM2 at moderate strength.2 The variant has been reported as Pathogenic in ClinVar (Variation ID: 3339587) by a reputable clinical diagnostic laboratory (LabCorp, SCV005204398), meeting PP5 at supporting strength.3 No variant-specific functional data, de novo observations, segregation data, or case-control prevalence data were identified in any reviewed publication. All six ClinVar-cited PMIDs discuss STAG2 and cohesin mutations at the gene level in myeloid malignancies without mentioning NM_001042749.1:c.1018-1G>A.4 The combination of 1 very strong (PVS1), 1 moderate (PM2), and 1 supporting (PP5) criterion meets the generic ACMG/AMP 2015 threshold for Pathogenic classification under the rule: 1 Very Strong + 1 Moderate + ≥1 Supporting.5
STAG2
Final classification
Pathogenic
STAG2 c.1018-1G>A · p.?
STAG2
NM_001042749.1:c.1018-1G>A is a canonical splice acceptor variant in STAG2, where loss of function is a known disease mechanism. The variant is assigned PVS1 at very strong strength under ClinGen SVI PVS1 recommendations (PMC6185798), supported by SpliceAI delta 1.00 predicting abolition of the canonical 3' acceptor site.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 moderate, PP5 supporting; combination = 1 very strong + 1 moderate + 1 supporting, which maps to Pathogenic.
Classification rationale
PVS1PM2PP5
Pathogenic
STAG2 c.1018-1G>A
PVS1 + PM2 + PP5
→
Pathogenic
1
pvs1_variant_assessmentpvs1_gene_contextpvs1_generic_framework ↗spliceai ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_001042749.1 · variants mapped to exon structure
STAG2
NM_001042749.1
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 17 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
review
Pathogenic
NM_001042749.1:c.1018-1G>A is a canonical splice acceptor variant (c.1018-1G>A, intron 11) predicted to disrupt normal splicing with SpliceAI delta score 1.00 (acceptor loss 1.0). Under ClinGen SVI PVS1 recommendations (PMC6185798), canonical ±1,2 splice variants are eligible for PVS1 at very strong strength when loss of function is an established disease mechanism for the gene. STAG2 germline LoF mechanism is supported by targeted literature review identifying multiple publications linking STAG2 loss of function to human disease.
Canonical splice acceptor variant at intron 11 (c.1018-1G>A)SpliceAI max delta 1.00 (DS_AL=1.0DS_AG=0.8)
✓
PM2
moderate
Pathogenic
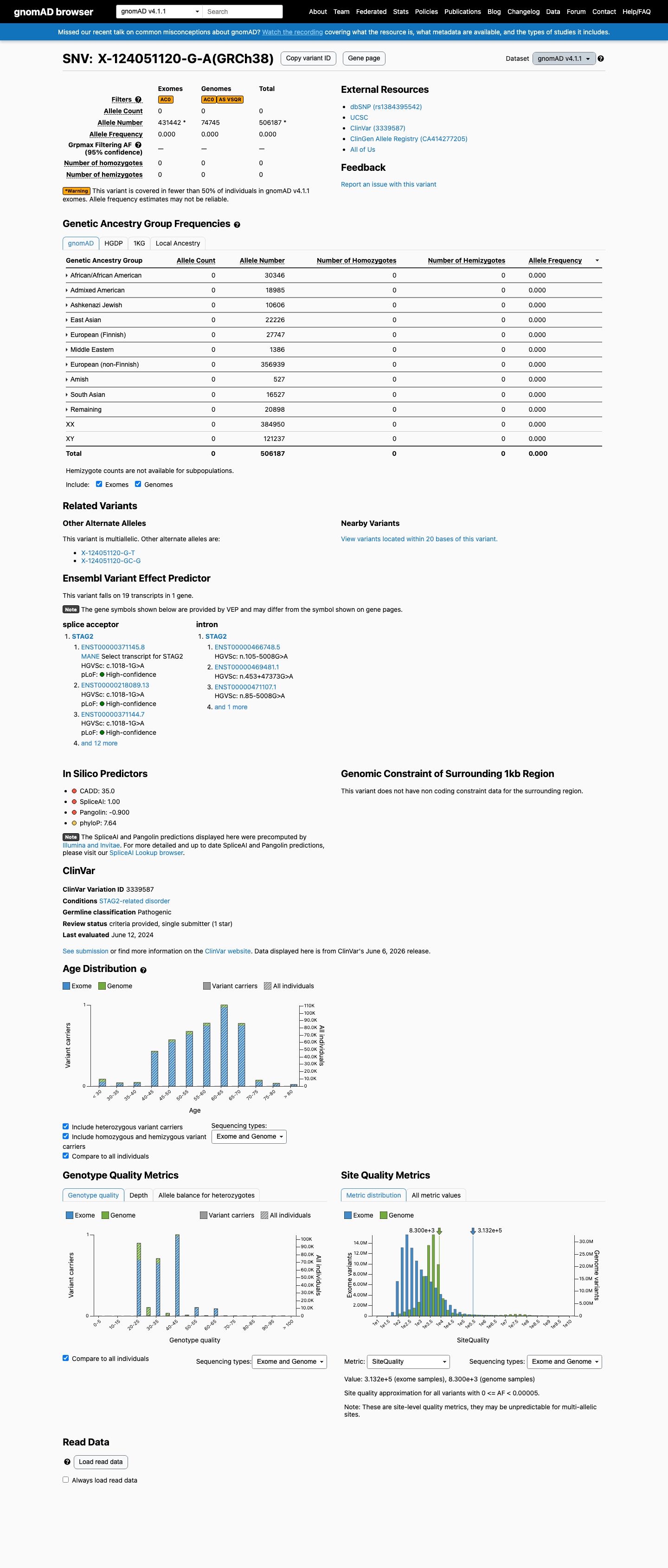
This variant is completely absent from gnomAD v2.1 and v4.1 (0/506,187 alleles across all populations). Allele frequency of 0% is well below the 0.1% PM2 threshold in all subpopulations, including the highest-observed African/African American population (0/30,346 alleles).
gnomAD v2.1: absentgnomAD v4.1: 0/506187 total alleles (AF=0.0)
✓
PP5
supporting
Pathogenic
This variant has been reported as Pathogenic in ClinVar (Variation ID: 3339587) by Women's Health and Genetics/Laboratory Corporation of America, LabCorp (SCV005204398), a reputable clinical diagnostic laboratory. Although the submission is from a single submitter with criteria provided, the classification from a CLIA-certified laboratory constitutes a reputable source report under ACMG/AMP PP5.
ClinVar Variation ID 3339587classified as PathogenicSubmitter: LabCorp (SCV005204398)
Assessed · not applied
Pathogenic
PS2
No de novo data available for this variant in any reviewed source.
PS3
No variant-specific functional data identified.
PS4
Insufficient variant-specific prevalence data in affected individuals.
PM1
No mutational hotspot at this position.
PM6
No de novo data available for this variant in any reviewed source.
PP1
No cosegregation data available for this variant.
PP4
No variant-specific phenotype or family history data available.
Benign
BA1
Allele frequency is 0% in gnomAD v4.1 (0/506,187 alleles), far below the 1% BA1 threshold.
BS1
Allele frequency is 0% in gnomAD v4.1, far below the 0.3% BS1 threshold.
BS2
No data on observation of this variant in healthy adult individuals available.
BS3
No functional studies demonstrating a benign or neutral effect for this variant exist in the available literature.
BS4
No cosegregation data available to assess lack of segregation with disease.
BP2
No data on observation of this variant in trans with a known pathogenic variant.
BP4
Multiple lines of computational evidence predict a deleterious effect.
BP5
No data on an alternate molecular cause for the phenotype in a case harboring this variant.
BP6
ClinVar reports this variant as Pathogenic (Variation ID: 3339587), not as benign.
BP7
This is a canonical splice acceptor variant (c.1018-1G>A) with SpliceAI max delta 1.00, predicting strong splice disruption.
N/A · 6
PS1 · PM5 · PP2 · PP3 · BP1 · BP3
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0; MAF= 0.00000%, 0/506187 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/30346 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / 506,187
0 hom
0 hom
Not observed in any ancestry group.
+ 10 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
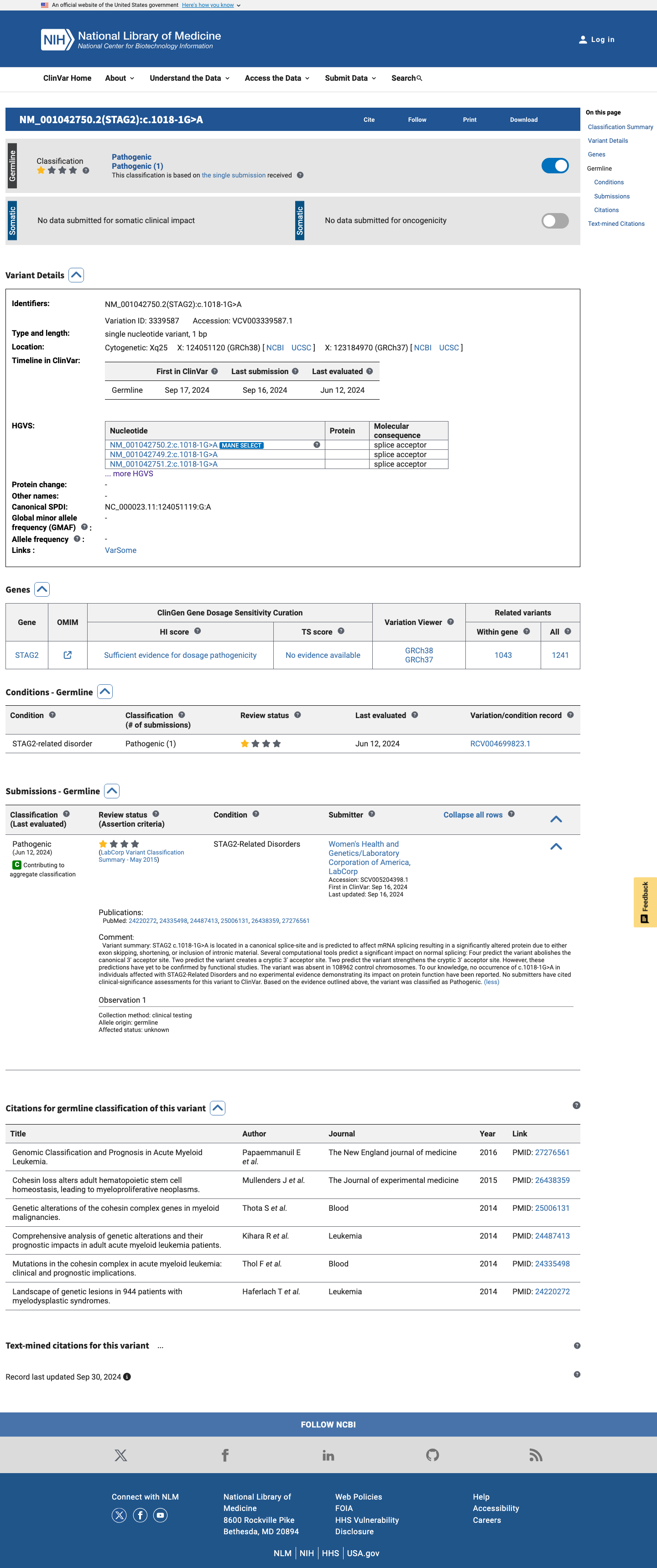
ClinVar
This variant has been reported in ClinVar as Pathogenic (1 clinical laboratory). (ClinVarID = 3339587)
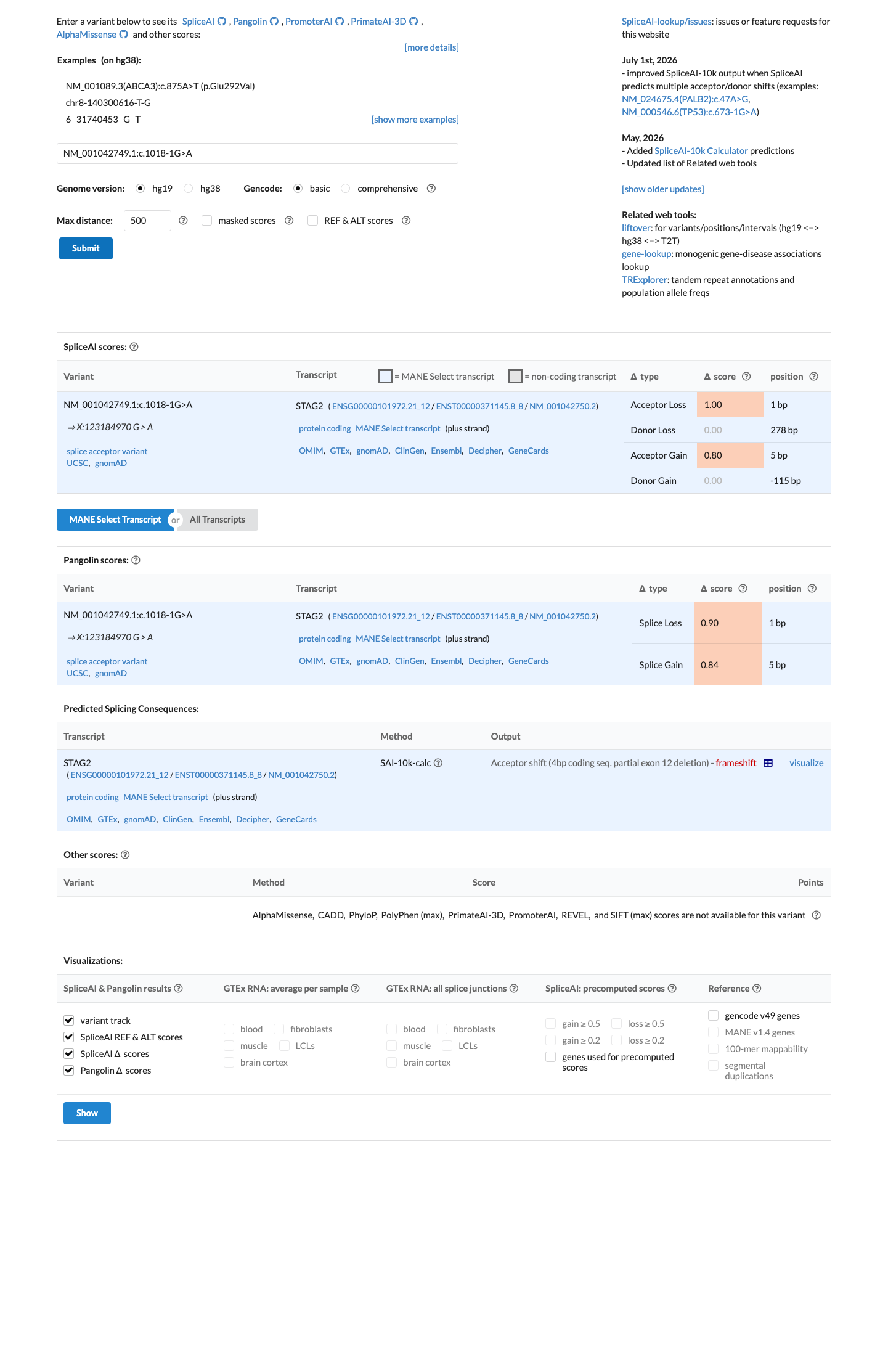
In silico
SpliceAI predicts possible splice impact for this variant (max delta score = 1.00). BayesDel score = 0.297619.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
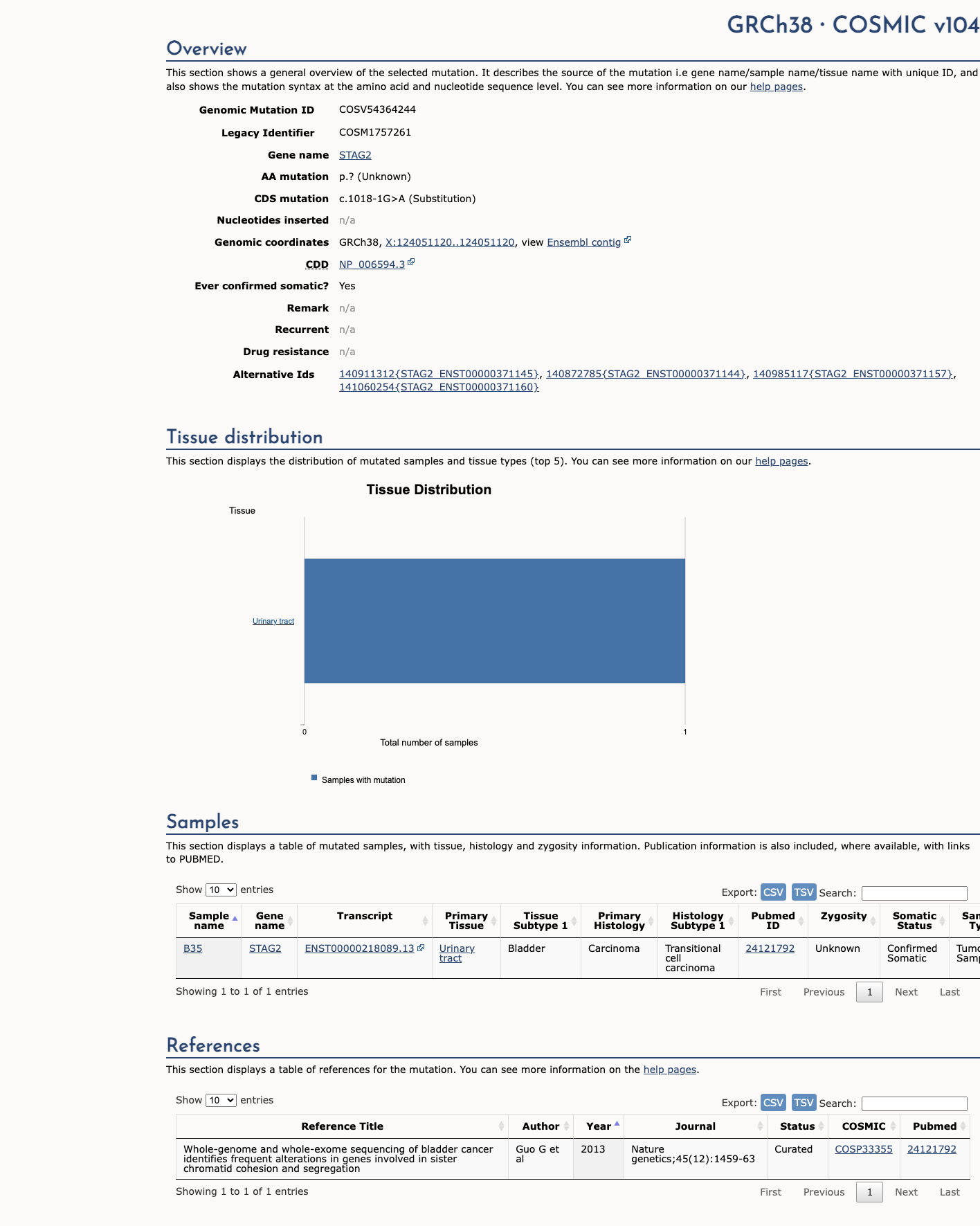
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV54364244, n = 1 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
24487413 ↗
Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients.
CLINVAR
26438359 ↗
Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms.
CLINVAR
24335498 ↗
Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications.
CLINVAR