NM_005228.4:c.2146A>G (p.Lys716Glu) is a missense variant in EGFR exon 18, encoding part of the kinase domain ATP-binding pocket. The variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada (PM2_supporting).1 In silico predictions are indeterminate: REVEL score 0.426 and BayesDel score 0.075 fall between benign and pathogenic thresholds; SpliceAI predicts no splicing impact (max delta 0.00). Neither PP3 nor BP4 is met.2 The variant has been reported once in a somatic context in a patient with lung adenocarcinoma enrolled in a study of non-classical EGFR mutations; p.Lys716Glu was classified as a resistant mutation to afatinib (progressive disease, PFS 1.1 months) (PMID:28676220). This observation does not meet PS3 or PS4 thresholds in a germline variant interpretation context.3 No experimental functional data, no segregation data, no de novo reports, and no pathogenic ClinVar classifications exist for this variant. ClinVar reports Uncertain significance from two clinical laboratories.4 With a single supporting-level criterion (PM2_supporting) and no other criteria met in either the pathogenic or benign direction, this variant is classified as a Variant of Uncertain Significance (VUS) under generic ACMG/AMP 2015 combination rules.5
EGFR
Final classification
VUS
EGFR c.2146A>G · p.Lys716Glu
EGFR
NM_005228.4:c.2146A>G (p.Lys716Glu) is a missense variant in EGFR exon 18, encoding part of the kinase domain ATP-binding pocket. The variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada (PM2_supporting).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting; combination = 1 supporting, which maps to VUS.
Classification rationale
PM2
VUS
EGFR c.2146A>G
PM2
→
VUS
2
revelbayesdelspliceai ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_005228.4 · variants mapped to exon structure
EGFR
NM_005228.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 22 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_005228.4:c.2146A>G is absent from gnomAD v2.1, gnomAD v4.1, and gnomAD-Canada v1.0. Allele frequency is 0% across all population databases, meeting the PM2 threshold for variants absent or present at extremely low frequency (<0.1%) in large population cohorts.
Absent from gnomAD v2.1 (AC=0).Absent from gnomAD v4.1 (AC=0).Absent from gnomAD-Canada v1.0 (AC=0).
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant at NM_005228.4 nucleotide position c.2146 with a different nucleotide change leading to the same amino acid substitution (p.Lys716Glu) has been identified in ClinVar or the literature.
PS2
No de novo occurrence has been documented for NM_005228.4:c.2146A>G in the available literature or case materials.
PS3
No experimental functional data (biochemical, cellular, or animal model) exists for p.Lys716Glu.
PS4
The variant has been observed in one patient in PMID:28676220 in a somatic NSCLC context and once in COSMIC (COSV104369001).
PM1
The variant p.Lys716Glu is located in EGFR exon 18, which encodes part of the ATP-binding pocket within the kinase domain — a critical functional domain.
PM6
No de novo germline observation has been documented for this variant.
PP1
No segregation data are available for this variant.
PP2
HCI prior probability is not available for EGFR (gene not supported by the HCI prior dataset).
PP3
In silico predictions are indeterminate: REVEL score is 0.426 (below the 0.5 threshold for deleterious prediction), BayesDel score is 0.075 (below typical deleterious thresholds), and SpliceAI predicts no splicing impact (max delta = 0.00).
PP4
No patient phenotype information is available for this variant in a germline context.
PP5
No reputable source has classified this variant as pathogenic.
Benign
BA1
The variant is absent from all gnomAD population databases (v2.1, v4.1, Canada).
BS1
The variant is absent from all gnomAD population databases.
BS2
The variant has not been observed in healthy adult controls.
BS3
No functional studies demonstrating a benign effect for p.Lys716Glu exist.
BS4
No segregation data are available for this variant.
BP1
NM_005228.4:c.2146A>G is a missense variant, not a truncating variant.
BP2
No co-occurrence data with a known pathogenic variant in EGFR are available.
BP4
In silico predictions are indeterminate: REVEL score 0.426 and BayesDel score 0.075 fall between benign and pathogenic thresholds.
BP5
No evidence that this variant is found in a case with an alternate molecular basis for disease.
BP6
No reputable source has classified this variant as benign.
BP7
NM_005228.4:c.2146A>G is a missense variant (p.Lys716Glu), not a synonymous variant.
N/A · 2
PVS1 · PM5
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
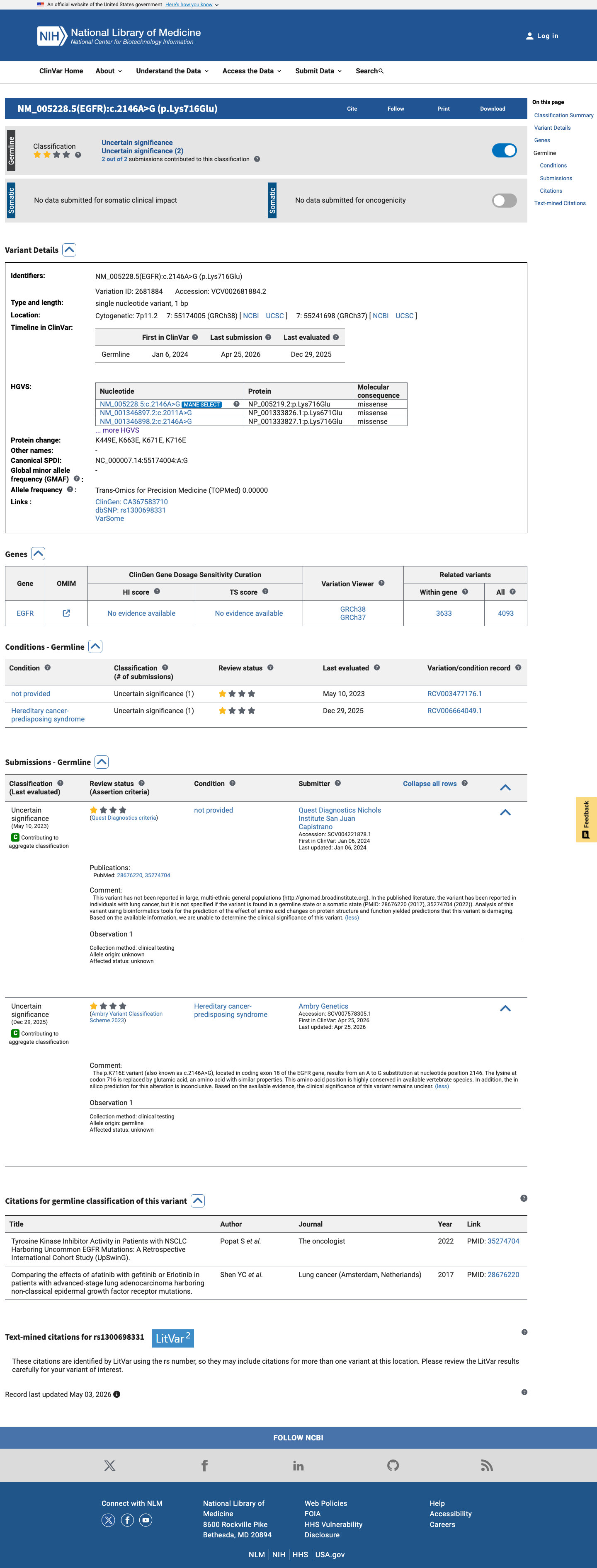
ClinVar
This variant has been reported in ClinVar as Uncertain significance (2 clinical laboratories). (ClinVarID = 2681884)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.426. BayesDel score = 0.0749666.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. EGFR, a receptor tyrosine kinase, is altered by amplification and/or mutation in lung and brain cancers among others.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV104369001, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
35274704 ↗
Tyrosine Kinase Inhibitor Activity in Patients with NSCLC Harboring Uncommon EGFR Mutations: A Retrospective International Cohort Study (UpSwinG).
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28676220 ↗
Comparing the effects of afatinib with gefitinib or Erlotinib in patients with advanced-stage lung adenocarcinoma harboring non-classical epidermal growth factor receptor mutations.
CLINVAR