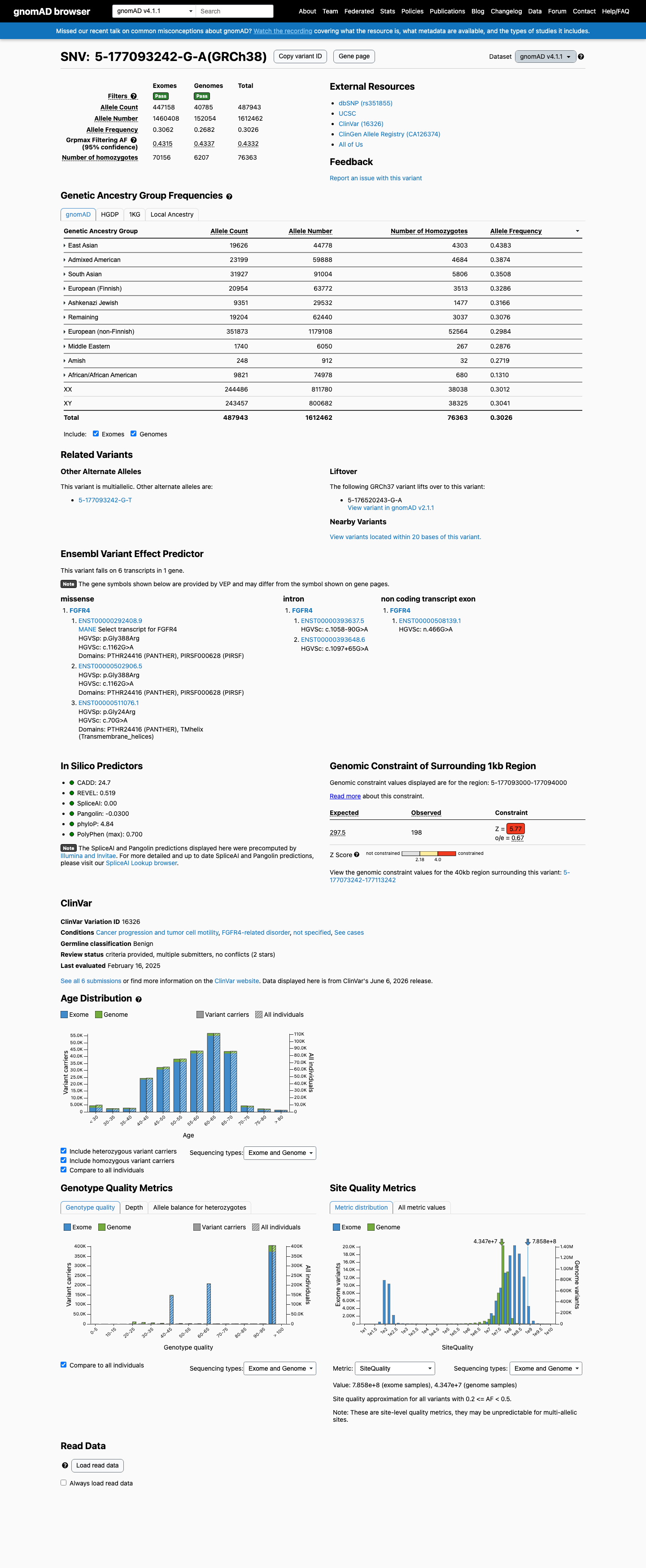
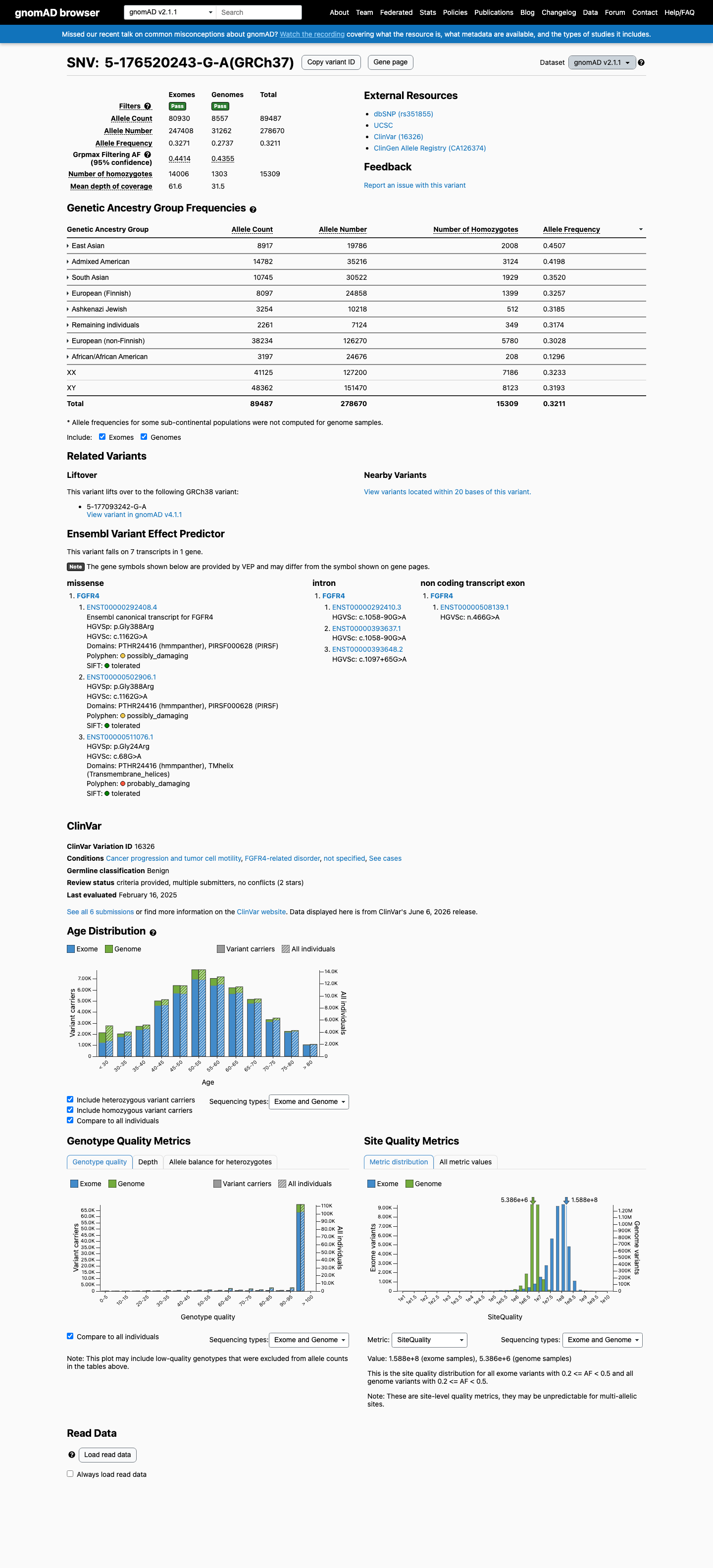
FGFR4 c.1162G>A (p.Gly388Arg) is an extremely common polymorphism with a global allele frequency of 32.1% in gnomAD v2.1 (89,487/278,670 alleles, 15,309 homozygotes) and 30.3% in gnomAD v4.1 (487,943/1,612,462 alleles, 76,363 homozygotes), meeting BA1 (stand-alone benign).1 The variant frequency far exceeds the threshold for any rare monogenic disorder (BS1 threshold >0.3%), with the highest subpopulation frequency of 45.1% in East Asians.2 The variant has been observed in the homozygous state in 15,309 individuals in gnomAD v2.1, consistent with Hardy-Weinberg equilibrium for a common benign polymorphism and incompatible with a fully penetrant pathogenic variant (BS2).3 FGFR4 germline disease is associated with loss-of-function; this is a missense variant not expected to recapitulate the LoF disease mechanism (BP1).4 Multiple in silico predictors are consistent with a benign interpretation: BayesDel score 0.0305 (benign), REVEL 0.519 (neutral), and SpliceAI max delta 0.02 (no splicing impact) (BP4).5 ClinVar classifies this variant as Benign based on submissions from 3 clinical laboratories (ClinVar VariationID 16326) (BP6).6 Functional studies demonstrate FGFR4 G388R has gain-of-function effects (STAT3 binding, increased cancer cell motility) in cancer models, but these do not establish pathogenicity for a monogenic germline disorder and are superseded by the overwhelming population frequency evidence.7
FGFR4
Final classification
Benign
FGFR4 c.1162G>A · p.Gly388Arg
FGFR4
FGFR4 c.1162G>A (p.Gly388Arg) is an extremely common polymorphism with a global allele frequency of 32.1% in gnomAD v2.1 (89,487/278,670 alleles, 15,309 homozygotes) and 30.3% in gnomAD v4.1 (487,943/1,612,462 alleles, 76,363 homozygotes), meeting BA1 (stand-alone benign).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 strong benign, BP1 supporting benign, BP4 supporting benign, BP6 supporting benign; combination = 1 stand-alone benign + 2 strong benign + 3 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BS2BP1BP4BP6
Benign
FGFR4 c.1162G>A
BA1 + BS1 + BS2 + BP1 + BP4 + BP6
→
Benign
4
pvs1_gene_context
5
revelbayesdelspliceai ↗
Gene diagram
· NM_213647.2 · variants mapped to exon structure
FGFR4
NM_213647.2
Fetching transcript structure from UCSC…
Applied criteria · 6 applied · 9 assessed
Applied · 6
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
Extremely high population frequency: gnomAD v2.1 AF=32.1% (89,487/278,670 alleles, 15,309 homozygotes) and gnomAD v4.1 AF=30.3% (487,943/1,612,462 alleles, 76,363 homozygotes). Far exceeds the BA1 threshold of >1%.
gnomAD v2.1: 89487/278670 alleles
✓
BS1
strong
Benign
Variant frequency (gnomAD AF=32.1%) far exceeds the threshold for any rare monogenic disorder (BS1 threshold >0.3%), and is inconsistent with a highly penetrant pathogenic role.
Global AF=32.1% (v2.1) and 30.3% (v4.1)orders of magnitude above the BS1 threshold of >0.3%.Observed across all major population groups with AF ranging from 12.9% (African) to 45.1% (East Asian).
✓
BS2
strong
Benign
Observed in homozygous state in 15,309 individuals in gnomAD v2.1 (76,363 in v4.1), demonstrating the variant is compatible with normal development and inconsistent with a fully penetrant pathogenic variant causing a severe monogenic disorder.
gnomAD v2.1: 15309 homozygotes observed.gnomAD v4.1: 76
✓
BP1
supporting
Benign
Missense variant in FGFR4, a gene for which loss-of-function is the documented disease mechanism in germline FGFR4-related disorders (familial pituitary adenomas). Truncating variants, not missense changes, are the primarily established pathogenic variant type.
FGFR4 germline disease mechanism is loss-of-function (supported by PMID:26186299 and other sources).Missense variants in a primarily LoF disease gene are less likely to be pathogenic.
✓
BP4
supporting
Benign
Multiple in silico predictors suggest no deleterious impact: BayesDel score=0.0305 (benign), REVEL=0.519 (borderline/neutral), SpliceAI max delta=0.02 (no splicing impact).
BayesDel: 0.0305 (strongly benign).REVEL: 0.519 (neutralwell below pathogenic threshold of ~0.75).
✓
BP6
supporting
Benign
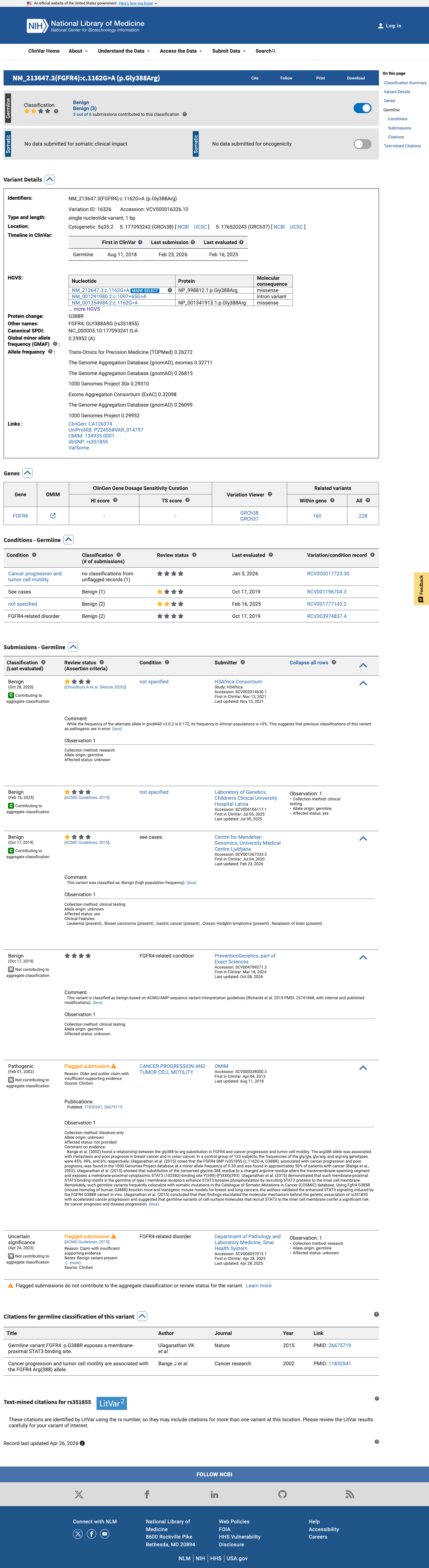
ClinVar classifies this variant as Benign based on submissions from 3 clinical laboratories (ClinVar VariationID 16326).
ClinVar VariationID 16326: Benign classificationcriteria provided (single submitter tier).3 clinical laboratories independently classify as Benign.
Assessed · not applied
Pathogenic
PS1
No alternative nucleotide change at codon 388 resulting in the same amino acid substitution (p.Gly388Arg) has been reported as pathogenic in ClinVar.
PS3
Functional studies demonstrate that FGFR4 p.Gly388Arg alters receptor biology (STAT3 binding site exposure, increased cancer cell motility), but these are gain-of-function effects in cancer models rather than evidence of a deleterious effect causing a monogenic germline disorder.
PS4
No case-control data comparing affected versus unaffected individuals for a specific FGFR4-related monogenic germline disorder are available.
PM1
The variant lies in the transmembrane domain of FGFR4 but is not located in a statistically significant mutational hotspot (cancerhotspots.org: not significant).
PM2
Variant is extremely common in population databases (gnomAD v2.1 AF=32.1%, v4.1 AF=30.3%), far exceeding the PM2 threshold of <0.1%.
PP2
FGFR4 does not have a low rate of benign missense variation; this variant itself is a common polymorphism present in over 30% of the general population.
PP3
Multiple in silico predictors do not support a deleterious effect: BayesDel score=0.0305 (benign), REVEL=0.519 (borderline/neutral), SpliceAI max delta=0.02 (no splicing impact).
PP5
ClinVar consensus is Benign (3 clinical laboratories).
Benign
BS3
Published functional studies (PMID:11830541, PMID:26675719) demonstrate that FGFR4 p.Gly388Arg has biological activity (STAT3 binding, altered cancer cell motility) rather than no effect.
N/A · 11
PVS1 · PS2 · PM5 · PM6 · PP1 · PP4 · BS4 · BP2 · BP3 · BP5 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.302607; MAF= 30.26074%, 487943/1612462 alleles, homozygotes = 76363) and has highest observed frequency in the East Asian population (AF= 0.438296; MAF= 43.82956%, 19626/44778 alleles, homozygotes = 4303); grpmax FAF= 0.433162.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.321122; MAF= 32.11218%, 89487/278670 alleles, homozygotes = 15309) and has highest observed frequency in the East Asian population (AF= 0.450672; MAF= 45.06722%, 8917/19786 alleles, homozygotes = 2008); grpmax FAF= 0.441438.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.3066724625081504, 5644/18404 alleles, homozygotes = 883).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
30%
· 487943 / 1,612,462
76363 hom · FAF 43%
76363 hom · FAF 43%
East Asian 19626 / 44,778 |
44% 4303 hom |
Admixed American 23199 / 59,888 |
39% 4684 hom |
South Asian 31927 / 91,004 |
35% 5806 hom |
European (Finnish) 20954 / 63,772 |
33% 3513 hom |
Ashkenazi Jewish 9351 / 29,532 |
32% 1477 hom |
Remaining individuals 19204 / 62,440 |
31% 3037 hom |
European (non-Finnish) 351873 / 1,179,108 |
30% 52564 hom |
Middle Eastern 1740 / 6,050 |
29% 267 hom |
Amish 248 / 912 |
27% 32 hom |
African/African American 9821 / 74,978 |
13% 680 hom |
gnomAD v2.1
32%
· 89487 / 278,670
15309 hom · FAF 44%
15309 hom · FAF 44%
East Asian 8917 / 19,786 |
45% 2008 hom |
Admixed American 14782 / 35,216 |
42% 3124 hom |
South Asian 10745 / 30,522 |
35% 1929 hom |
European (Finnish) 8097 / 24,858 |
33% 1399 hom |
Ashkenazi Jewish 3254 / 10,218 |
32% 512 hom |
Remaining individuals 2261 / 7,124 |
32% 349 hom |
European (non-Finnish) 38234 / 126,270 |
30% 5780 hom |
African/African American 3197 / 24,676 |
13% 208 hom |
gnomAD Canada 🇨🇦
31%
· 5644 / 18,404
883 hom · FAF 44%
883 hom · FAF 44%
indel · split
East Asian 636 / 1,338 |
48% 150 hom |
South Asian 495 / 1,360 |
36% 87 hom |
Latino/Admixed American 286 / 836 |
34% 48 hom |
Ashkenazi Jewish 256 / 832 |
31% 36 hom |
Remaining individuals 343 / 1,138 |
30% 54 hom |
European (non-Finnish) 3474 / 11,730 |
30% 498 hom |
Middle Eastern 31 / 142 |
22% 1 hom |
European (Finnish) 1 / 8 |
12% |
African/African American 122 / 1,020 |
12% 9 hom |
ClinVar
This variant has been reported in ClinVar as Benign (3 clinical laboratories). (ClinVarID = 16326)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). REVEL score = 0.519. BayesDel score = 0.0304758.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. FGFR4, a receptor tyrosine kinase, is altered by mutation, chromosomal rearrangement or amplification at low frequencies in various cancer types.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
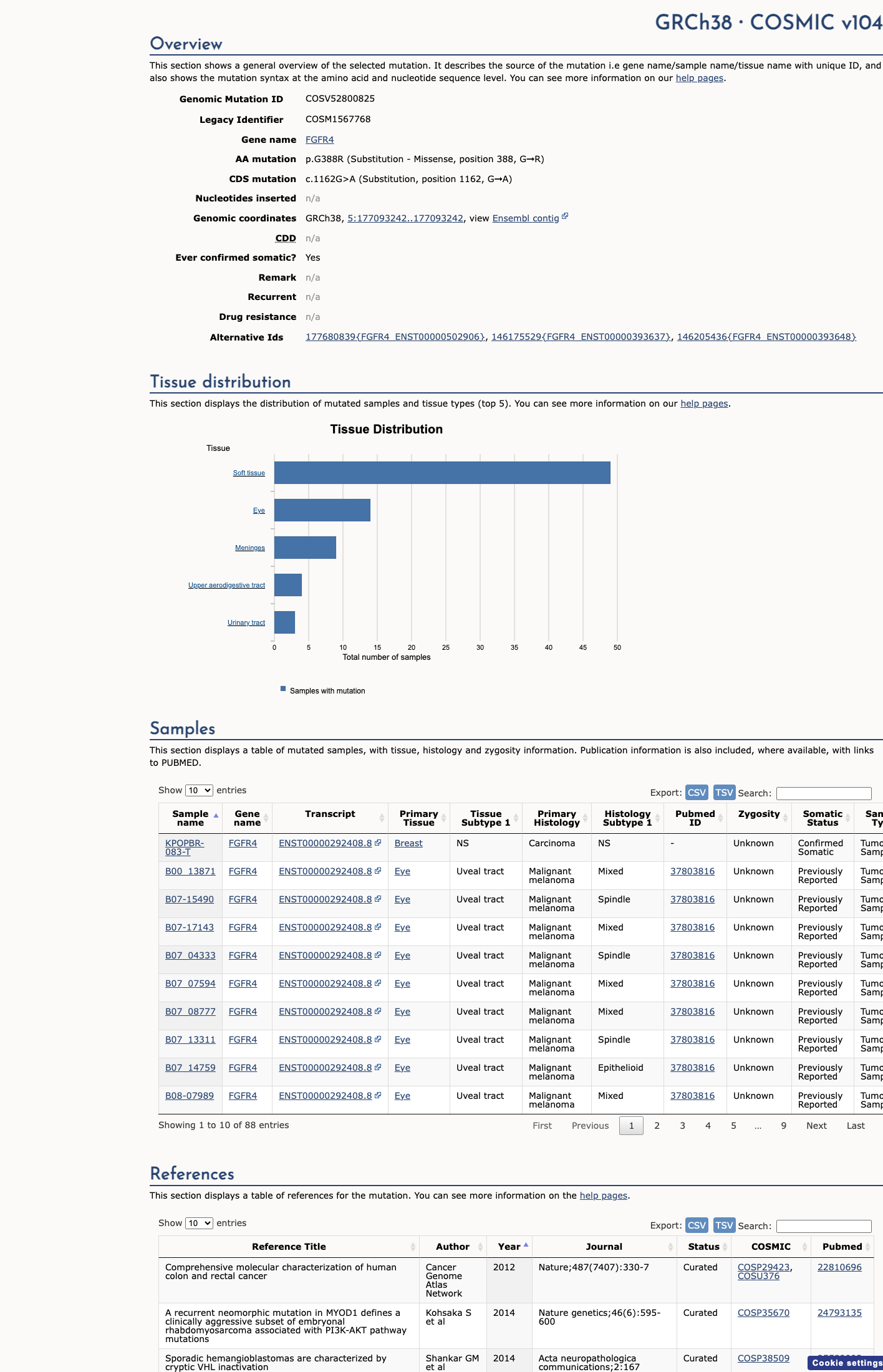
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV52800825, n = 88 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
11830541 ↗
Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26675719 ↗
Germline variant FGFR4  p.G388R exposes a membrane-proximal STAT3 binding site.
CLINVAR