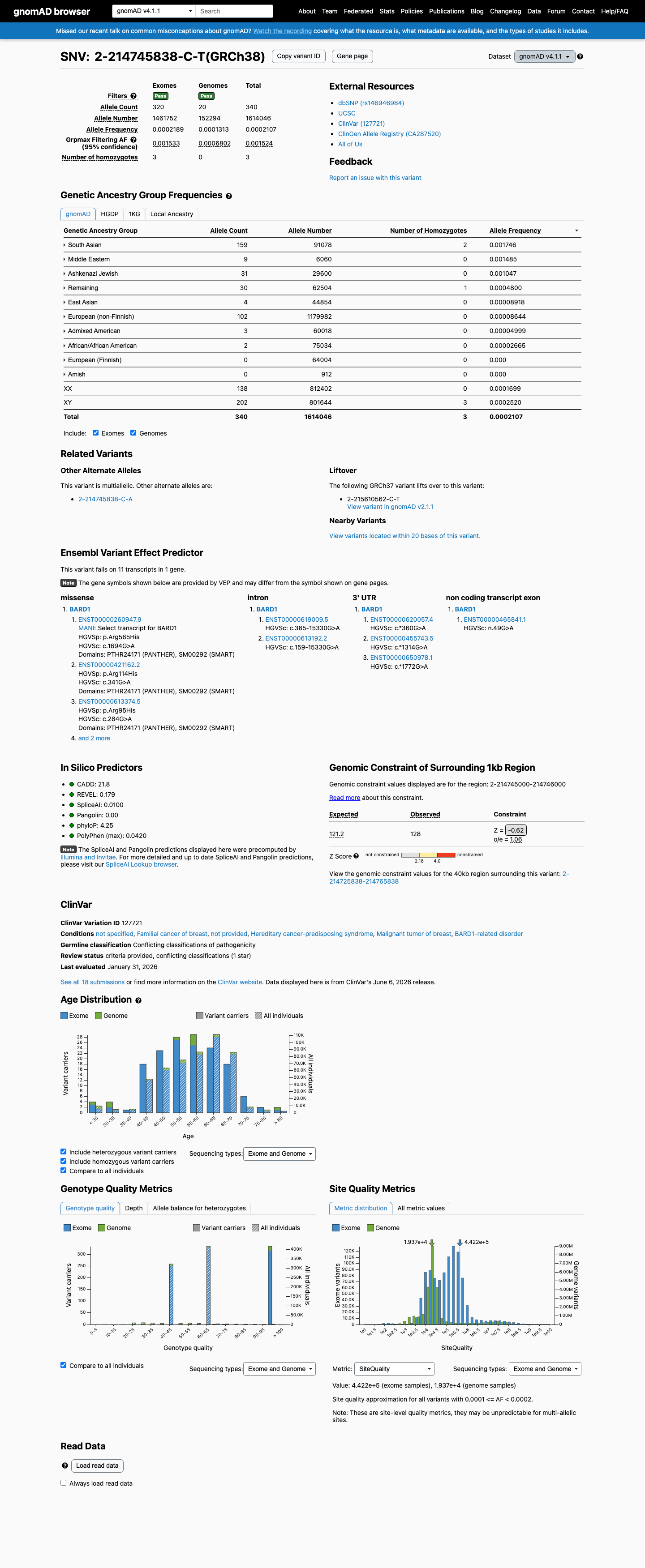
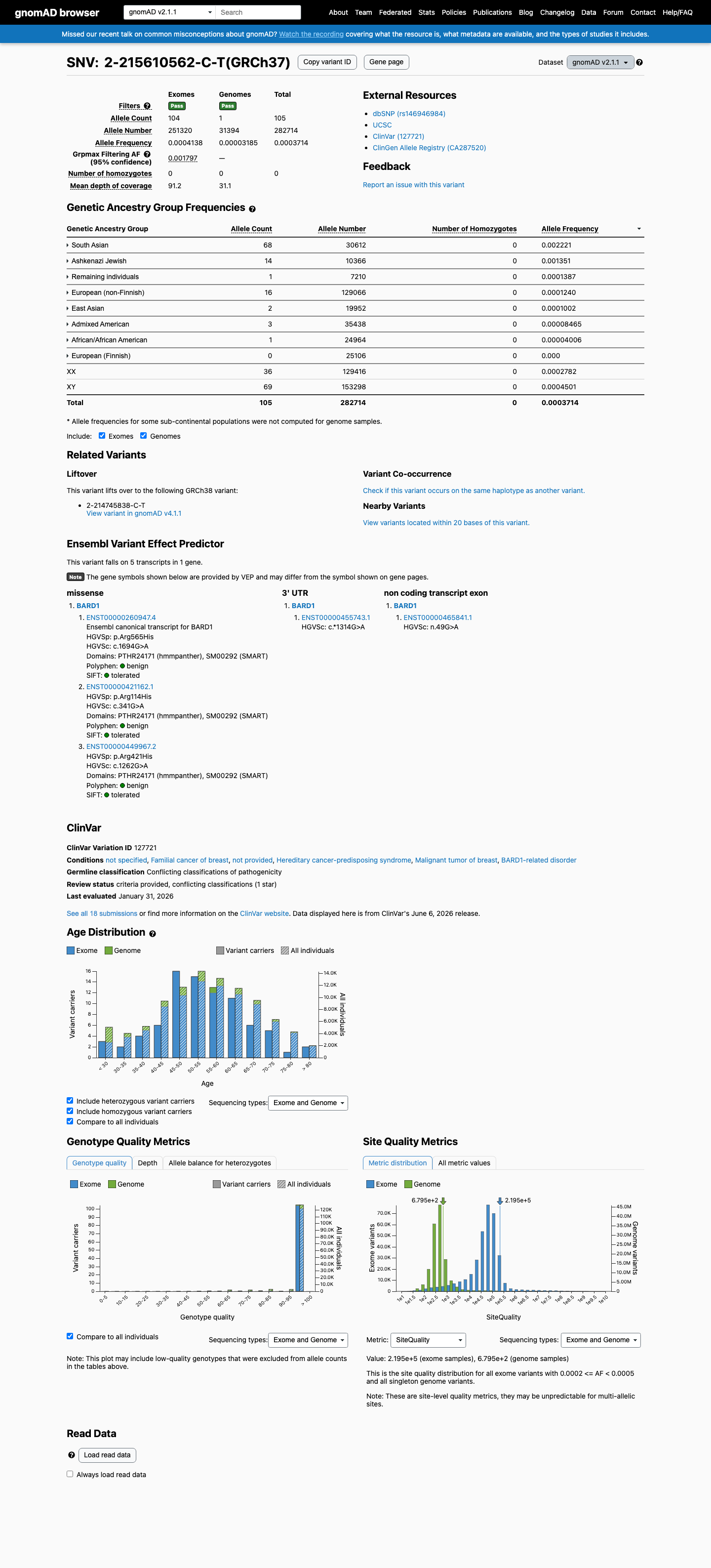
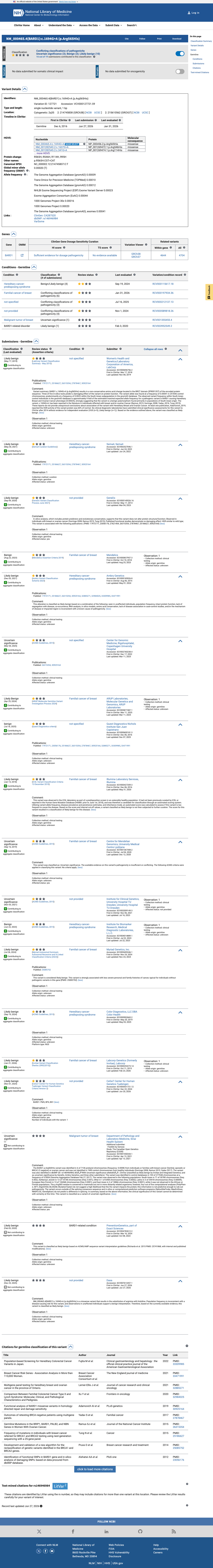
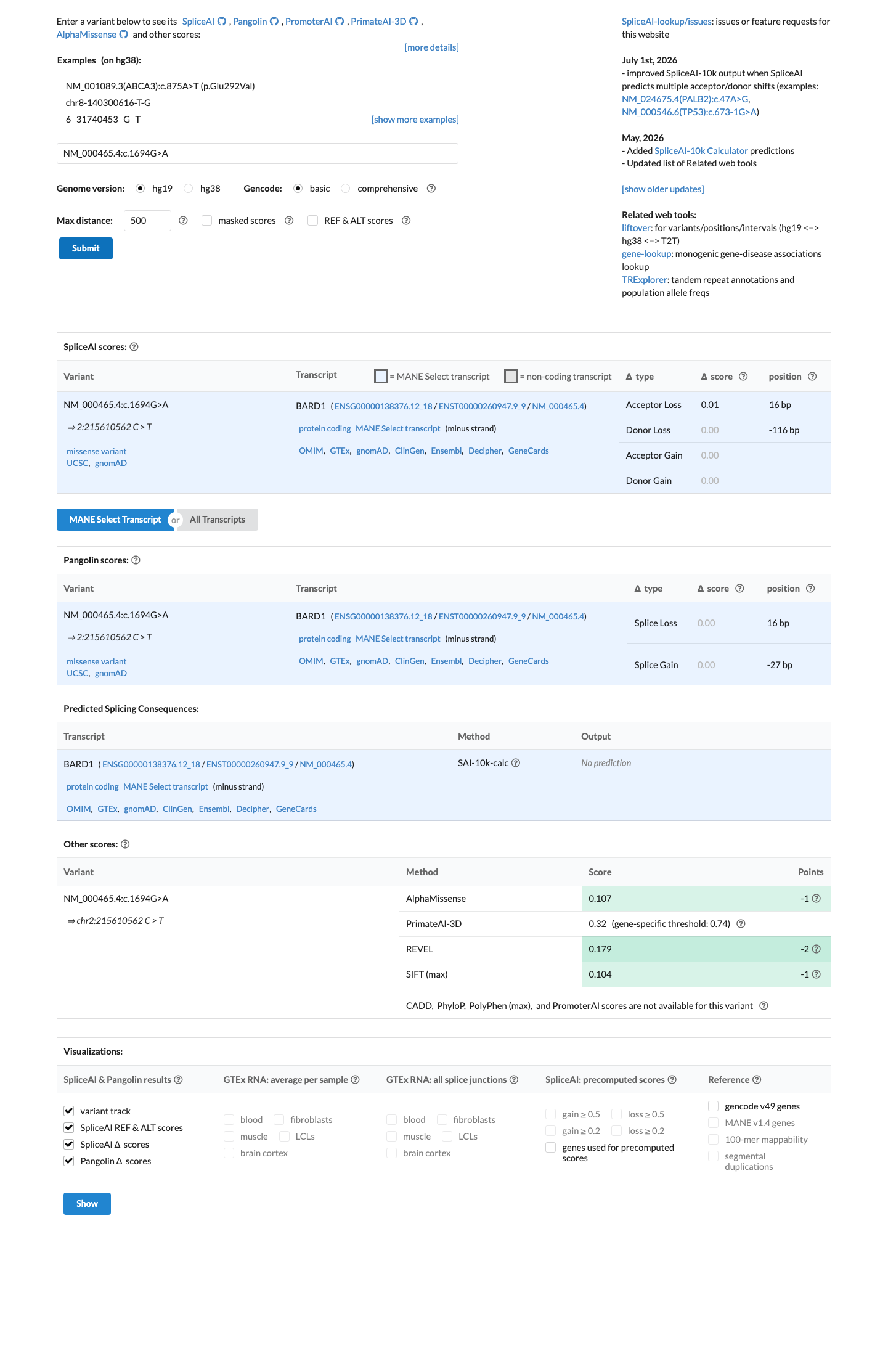
BARD1 c.1694G>A (p.Arg565His) is a missense variant in exon 8 located in the inter-domain region between the ankyrin repeat and BRCT domains.1 Direct functional testing in a validated homology-directed repair (HDR) assay demonstrated that BARD1 R565H is fully functional, with HDR activity comparable to wild-type. The variant was explicitly classified as 'functional in HDR' alongside known benign variants (PMID:30925164).2 This variant is present in gnomAD v2.1 at 105/282,714 alleles (AF=0.037%) and in gnomAD v4.1 at 340/1,614,046 alleles (AF=0.021%), with 3 homozygous individuals observed in v4.1. Homozygosity in a general population database is inconsistent with a highly penetrant pathogenic variant in a tumor suppressor gene.3 Multiple in silico tools support a benign effect: REVEL score 0.179, BayesDel score -0.24551, and SpliceAI max delta 0.01 (no splicing impact).4 ClinVar reports this variant as Likely benign based on submissions from 10 clinical laboratories, with an additional 2 laboratories reporting Benign and 4 reporting Uncertain significance (Variation ID 127721).5 The variant was observed in 1 of 354 sporadic breast cancer cases and 0 of 258 controls (PMID:17972171), which does not establish significant case enrichment. A single FCCTX colorectal cancer case carrying this variant has also been reported, but colorectal cancer is not within the primary BARD1-associated disease spectrum (PMID:32984025).6 No evidence of de novo occurrence, co-segregation with disease, or location within a critical functional domain or mutational hotspot was identified. Residue 565 lies in a linker region where variants tested in functional studies were generally proficient in DNA repair.7
BARD1
Final classification
Likely Benign
BARD1 c.1694G>A · p.Arg565His
BARD1
BARD1 c.1694G>A (p.Arg565His) is a missense variant in exon 8 located in the inter-domain region between the ankyrin repeat and BRCT domains.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS2 supporting benign, BS3 strong benign, BP4 supporting benign; combination = 1 strong benign + 2 supporting benign, which maps to Likely Benign.
Classification rationale
BS2BS3BP4
Likely Benign
BARD1 c.1694G>A
BS2 + BS3 + BP4
→
Likely Benign
4
revelbayesdelspliceai ↗
Gene diagram
· NM_000465.4 · variants mapped to exon structure
BARD1
NM_000465.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 19 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS2
supporting
Benign
Observed in the homozygous state in 3 individuals in gnomAD v4.1, a general population database. BARD1 is a tumor suppressor gene where biallelic loss-of-function would be expected to have significant phenotypic consequences. The observation of homozygosity in presumably healthy population controls suggests the variant is tolerated even in the homozygous state.
gnomAD v4.1: 3 homozygous individuals observed among 340 total allele counts.Homozygosity in a general population database is inconsistent with a highly penetrant pathogenic variant.
✓
BS3
strong
Benign
Direct functional testing of BARD1 R565H in a validated homology-directed repair (HDR) assay demonstrated unequivocally normal function. Adamovich et al. (PMID:30925164) tested 76 BARD1 missense variants in a well-characterized HDR assay that accurately discriminates known pathogenic from known benign BARD1 variants. R565H was proficient in HDR with activity comparable to wild-type and was explicitly grouped with other functional variants that were interpreted as 'likely benign.' The HDR assay has been validated as predictive of BRCA1/BARD1 pathogenicity, with truncating variants and known pathogenic missense variants showing HDR deficiency while known benign variants remain functional.
HDR assay in HeLa-DR cells: BARD1 R565H showed normal HDR functioncomparable to wild-type.R565H was explicitly listed among variants 'which were functional in HDR' and grouped with likely benign variants.
✓
BP4
supporting
Benign
Multiple lines of computational evidence support a benign effect. REVEL score is 0.179 (well below damaging thresholds). BayesDel score is -0.24551 (negative, supporting benign). SpliceAI max delta score is 0.01 (no predicted splicing impact). The preponderance of in silico evidence from validated ensemble predictors supports a benign interpretation, consistent with the functional data showing normal HDR activity.
REVEL score: 0.179 (benign).BayesDel score: -0.24551 (negativesupporting benign).
Assessed · not applied
Pathogenic
PS1
No evidence of a different nucleotide change at codon 565 resulting in the same amino acid substitution (p.Arg565His) that has been classified as pathogenic.
PS2
No de novo observation with confirmed maternity and paternity has been reported for this variant in any reviewed publication or database.
PS3
Direct functional testing of BARD1 R565H in a validated homology-directed repair (HDR) assay demonstrated that the variant is functional and proficient in DNA repair, comparable to wild-type.
PS4
Observed in 1 of 354 sporadic breast cancer cases and 0 of 258 controls (PMID:17972171), which does not establish statistically significant enrichment.
PM1
Residue Arg565 lies in the inter-domain region between the ankyrin repeat domain (residues 427-525) and the first BRCT domain (residues 616-653).
PM2
Although total allele frequency in gnomAD v2.1 is 0.037% (below 0.1% PM2 threshold), the variant is present at 105 alleles in v2.1 and 340 alleles in v4.1 with 3 homozygous individuals observed in v4.1.
PM5
No known pathogenic missense variant at codon 565 with a different amino acid change has been established.
PM6
No de novo observation (maternity and paternity unconfirmed) has been reported for this variant in any reviewed source.
PP1
No co-segregation data are available.
PP2
BARD1 has a high rate of benign missense variation.
PP3
Multiple in silico tools predict a benign effect.
PP4
The variant has been observed in a colorectal cancer case (FCCTX, PMID:32984025) and a sporadic breast cancer case (PMID:17972171).
PP5
ClinVar review status for this variant is 'criteria provided, single submitter' (1-star).
Benign
BA1
Allele frequency in gnomAD v2.1 is 0.037% (total) with maximum subpopulation frequency 0.22% (South Asian).
BS1
Allele frequency in gnomAD v2.1 is 0.037% (total), which is below the 0.3% BS1 threshold (non-VCEP).
BS4
No co-segregation data are available for this variant.
BP2
No data are available regarding observation of this variant in trans with a known pathogenic variant in BARD1.
BP5
No data are available demonstrating that a case carrying this variant has an alternate molecular basis for disease.
BP6
Although ClinVar reports this variant as Likely benign with strong clinical laboratory consensus (10 labs reporting Likely benign, 2 reporting Benign, 4 reporting VUS), the review status is 'criteria provided, single submitter' (1-star).
N/A · 6
PVS1 · PM3 · PM4 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000210651; MAF= 0.02107%, 340/1614046 alleles, homozygotes = 3) and has highest observed frequency in the South Asian population (AF= 0.00174576; MAF= 0.17458%, 159/91078 alleles, homozygotes = 2); grpmax FAF= 0.00152378.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.0003714; MAF= 0.03714%, 105/282714 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.00222135; MAF= 0.22214%, 68/30612 alleles, homozygotes = 0); grpmax FAF= 0.00179691.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.021%
· 340 / 1,614,046
3 hom · FAF 0.15%
3 hom · FAF 0.15%
South Asian 159 / 91,078 |
0.17% 2 hom |
Middle Eastern 9 / 6,060 |
0.15% |
Ashkenazi Jewish 31 / 29,600 |
0.1% |
Remaining individuals 30 / 62,504 |
0.048% 1 hom |
East Asian 4 / 44,854 |
0.0089% |
European (non-Finnish) 102 / 1,179,982 |
0.0086% |
Admixed American 3 / 60,018 |
0.005% |
African/African American 2 / 75,034 |
0.0027% |
+ 2 not observed (European (Finnish), Amish)
gnomAD v2.1
0.037%
· 105 / 282,714
0 hom · FAF 0.18%
0 hom · FAF 0.18%
South Asian 68 / 30,612 |
0.22% |
Ashkenazi Jewish 14 / 10,366 |
0.14% |
Remaining individuals 1 / 7,210 |
0.014% |
European (non-Finnish) 16 / 129,066 |
0.012% |
East Asian 2 / 19,952 |
0.01% |
Admixed American 3 / 35,438 |
0.0085% |
African/African American 1 / 24,964 |
0.004% |
+ 1 not observed (European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Likely benign (10 clinical laboratories) and as Uncertain significance (4 clinical laboratories) and as Benign (2 clinical laboratories) and as Likely Benign (1 clinical laboratory) and as benign (1 clinical laboratory). (ClinVarID = 127721)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.179. BayesDel score = -0.24551.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. BARD1, a tumor suppressor involved in the DNA damage response, is altered by mutation in breast and ovarian cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99640229, n = 3 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 8 further PMIDs triaged but not cited — see Sources & References.
Functional analysis of BARD1 missense variants in homology-directed repair and damage sensitivity.
Searched
c.1694G>Ap.Arg565HisR565HArg565
Found
BARD1 R565H was directly tested in a validated homology-directed repair (HDR) functional assay in HeLa-DR cells alongside 75 other BARD1 missense variants. R565H was proficient in HDR with activity comparable to wild-type and was explicitly grouped among 'functional in HDR' variants. The authors interpreted HDR-functional variants as 'likely benign,' noting that this interpretation is consistent with ClinVar classifications where known pathogenic variants are HDR-deficient and known benign variants are HDR-functional.
Variant
✓ Names this variant — characterised directly
Applied to
→BS3 supports · met
Why
Direct functional evidence of normal HDR function. Key evidence supporting BS3 (strong benign). Also evaluated for PS3 (not met, evidence contradicts damaging effect).
The variants V85L, R194K, I258T, N326S, R565H, and R641Q, which were functional in HDR, have conflicting reports of pathogenicity, with reports indicating they were VUS or likely benign. Since these variants were functional in the HDR assay, we would interpret such variants as likely benign.
Location Results section, 'Analysis of BARD1 variants in Homology-Directed Repair (HDR)'; Discussion section · Context Homology-directed repair (HDR) assay in HeLa-DR cells; endogenous BARD1 depleted by siRNA, variant BARD1 expressed from plasmid; flow cytometry readout of GFP-positive cells. · full text
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
17972171 ↗
BARD1 variants are not associated with breast cancer risk in Australian familial breast cancer.
CLINVAR
23056176 ↗
Identification of functional SNPs in BARD1 gene and in silico analysis of damaging SNPs: based on data procured from dbSNP database.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
32984025 ↗
Comparison Between Familial Colorectal Cancer Type X and Lynch Syndrome: Molecular, Clinical, and Pathological Characteristics and Pedigrees.
CLINVAR
24366376 ↗
Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR