NM_003579.4(RAD54L):c.888C>T (p.Asp296=) is a synonymous variant with no predicted impact on splicing (SpliceAI max delta 0.02), meeting BP7 at supporting strength.1 This variant has been reported in ClinVar as Likely benign by two clinical laboratories (ClinVarID 801488), meeting BP6 at supporting strength.2 REVEL, BayesDel, and HCI prior scores are not available for this variant. SpliceAI alone does not constitute multiple lines of computational evidence required for BP4. The variant is present in gnomAD v2.1 (103/282,688 alleles, AF=0.036%) and gnomAD v4.1 (655/1,613,818 alleles, AF=0.041%). The allele frequency is below the BS1 threshold (0.3%) and far below the BA1 threshold (5%). The variant is observed across multiple populations, which is inconsistent with PM2 absence.3 No functional studies, de novo observations, co-segregation data, case-control data, or phenotype information are available for this variant. All other assessed criteria are not met or not applicable. No CSPEC or VCEP framework exists for RAD54L. The generic ACMG/AMP 2015 classification framework (PMID:25741868) was applied.4 Two supporting benign criteria (BP6, BP7) are met. Per the generic ACMG/AMP combination rules, two supporting benign criteria classify a variant as Likely Benign.5
RAD54L
Final classification
Likely Benign
RAD54L c.888C>T · p.Asp296=
RAD54L
NM_003579.4(RAD54L):c.888C>T (p.Asp296=) is a synonymous variant with no predicted impact on splicing (SpliceAI max delta 0.02), meeting BP7 at supporting strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BP6 supporting benign, BP7 supporting benign; combination = 2 supporting benign, which maps to Likely Benign.
Classification rationale
BP6BP7
Likely Benign
RAD54L c.888C>T
BP6 + BP7
→
Likely Benign
4
generic_acmg_combination_rules
5
generic_acmg_combination_rules
Gene diagram
· NM_003579.4 · variants mapped to exon structure
RAD54L
NM_003579.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 18 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BP6
supporting
Benign
This variant is classified as Likely benign in ClinVar (ClinVarID 801488) by 2 clinical laboratories (Mendelics, Ambry Genetics). ClinVar is a reputable source for variant classification.
ClinVar Likely benign from 2 clinical laboratories (ClinVarID 801488)
✓
BP7
supporting
Benign
This is a synonymous variant (NM_003579.4:c.888C>T, p.Asp296=) with no predicted impact on splicing. SpliceAI predicts a maximum delta score of 0.02, indicating no significant alteration to splice consensus sequences or creation of new splice sites. The variant is present in gnomAD across multiple populations, inconsistent with high nucleotide conservation.
Synonymous variant p.(Asp296=)SpliceAI max delta 0.02 (no splicing impact)observed in gnomAD across populations
Assessed · not applied
Pathogenic
PS2
No de novo observation reported for this variant in ClinVar, literature, or any reviewed source.
PS3
No functional studies testing this exact variant or a systematically characterized range that includes c.888 were identified.
PS4
No case-control or prevalence data comparing this variant in affected versus unaffected individuals is available.
PM1
No statistically significant mutational hotspot at this residue was identified (cancerhotspots.org).
PM2
This variant is present in gnomAD v2.1 (103/282,688 alleles, AF=0.036%) and gnomAD v4.1 (655/1,613,818 alleles, AF=0.041%), including 0 homozygotes.
PM6
No de novo observation with or without confirmed parentage is reported for this variant.
PP1
No co-segregation data with disease in multiple affected family members is available for this variant.
PP3
No in silico tools predict a deleterious effect.
PP4
No patient phenotype or family history data specific to the disease gene is available for adjudication.
PP5
ClinVar classifies this variant as Likely benign, not pathogenic.
Benign
BA1
The highest observed allele frequency is 0.065% (gnomAD v2.1 European non-Finnish) and 0.099% (gnomAD v4.1 Middle Eastern), both far below the 5% threshold for BA1.
BS1
The global allele frequency is 0.036% (gnomAD v2.1) and 0.041% (gnomAD v4.1), below the 0.3% BS1 threshold for a non-VCEP assessment.
BS2
No data on observation of this variant in a healthy adult individual for a fully penetrant disorder is available.
BS3
No experimental functional studies demonstrating a benign effect for this variant were identified.
BS4
No evidence of lack of segregation with disease in affected family members is available.
BP2
No evidence that this variant has been observed in trans with a pathogenic variant in RAD54L.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact.
BP5
No evidence that this variant has been observed in a case with an alternate molecular basis for disease.
N/A · 5
PVS1 · PS1 · PM5 · PP2 · BP1
Research & evidence
Population frequency · supports benign
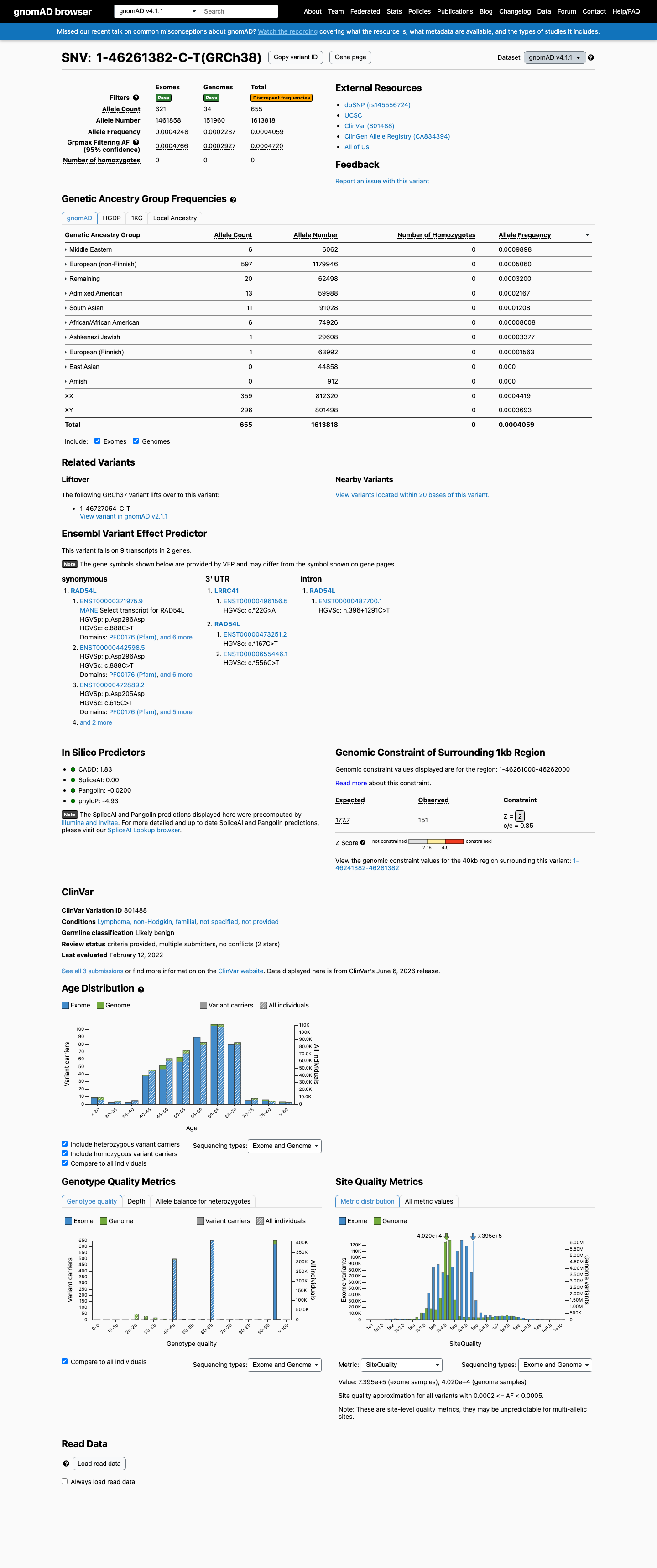
gnomAD v4.1
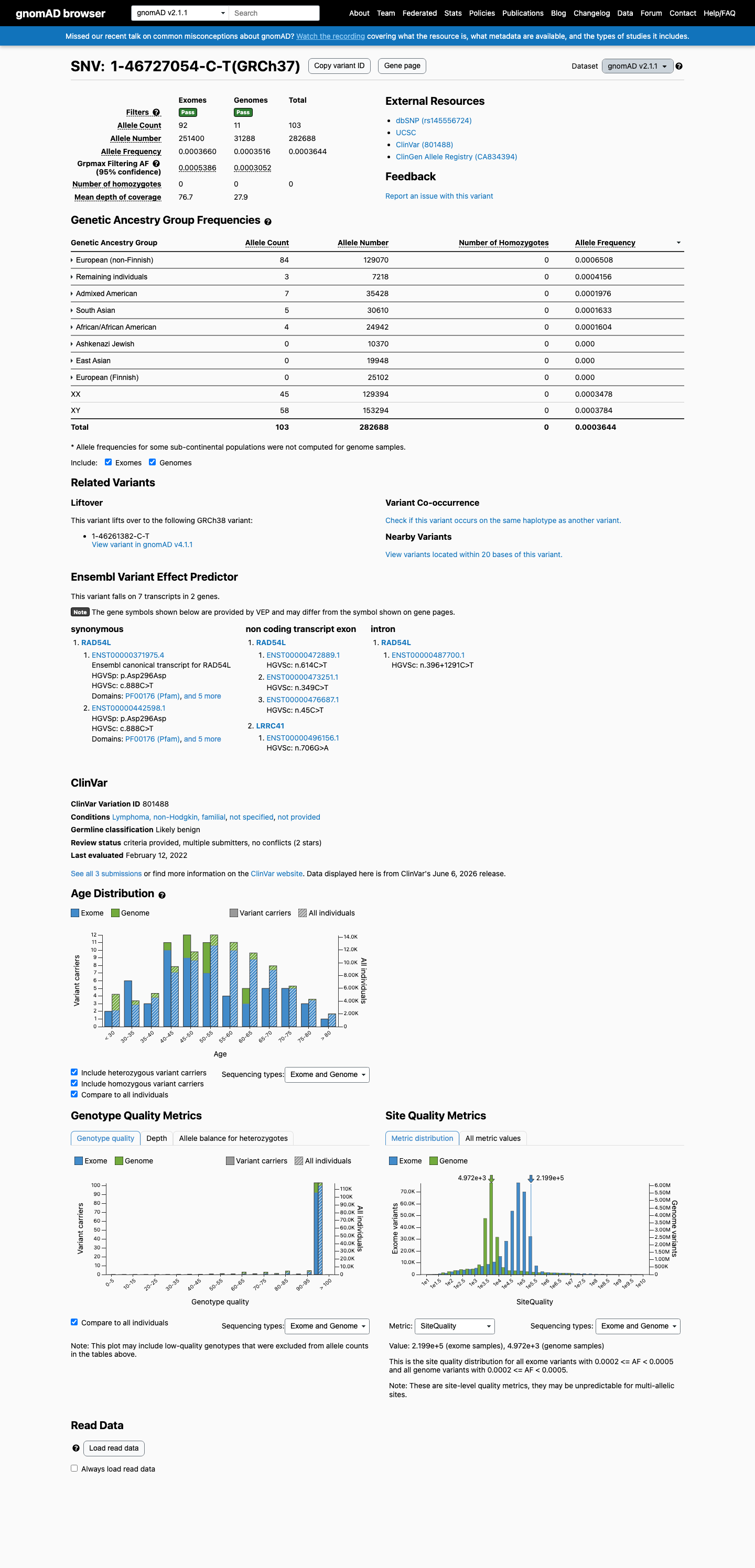
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00040587; MAF= 0.04059%, 655/1613818 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000989772; MAF= 0.09898%, 6/6062 alleles, homozygotes = 0); grpmax FAF= 0.00047202.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000364359; MAF= 0.03644%, 103/282688 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.00065081; MAF= 0.06508%, 84/129070 alleles, homozygotes = 0); grpmax FAF= 0.00053859.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0006515365403409708, 12/18418 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.041%
· 655 / 1,613,818
0 hom · FAF 0.047%
0 hom · FAF 0.047%
Middle Eastern 6 / 6,062 |
0.099% |
European (non-Finnish) 597 / 1,179,946 |
0.051% |
Remaining individuals 20 / 62,498 |
0.032% |
Admixed American 13 / 59,988 |
0.022% |
South Asian 11 / 91,028 |
0.012% |
African/African American 6 / 74,926 |
0.008% |
Ashkenazi Jewish 1 / 29,608 |
0.0034% |
European (Finnish) 1 / 63,992 |
0.0016% |
+ 2 not observed (Amish, East Asian)
gnomAD v2.1
0.036%
· 103 / 282,688
0 hom · FAF 0.054%
0 hom · FAF 0.054%
European (non-Finnish) 84 / 129,070 |
0.065% |
Remaining individuals 3 / 7,218 |
0.042% |
Admixed American 7 / 35,428 |
0.02% |
South Asian 5 / 30,610 |
0.016% |
African/African American 4 / 24,942 |
0.016% |
+ 3 not observed (Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
0.065%
· 12 / 18,418
0 hom · FAF 0.046%
0 hom · FAF 0.046%
Middle Eastern 1 / 144 |
0.69% |
Remaining individuals 1 / 1,138 |
0.088% |
European (non-Finnish) 10 / 11,738 |
0.085% |
+ 6 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
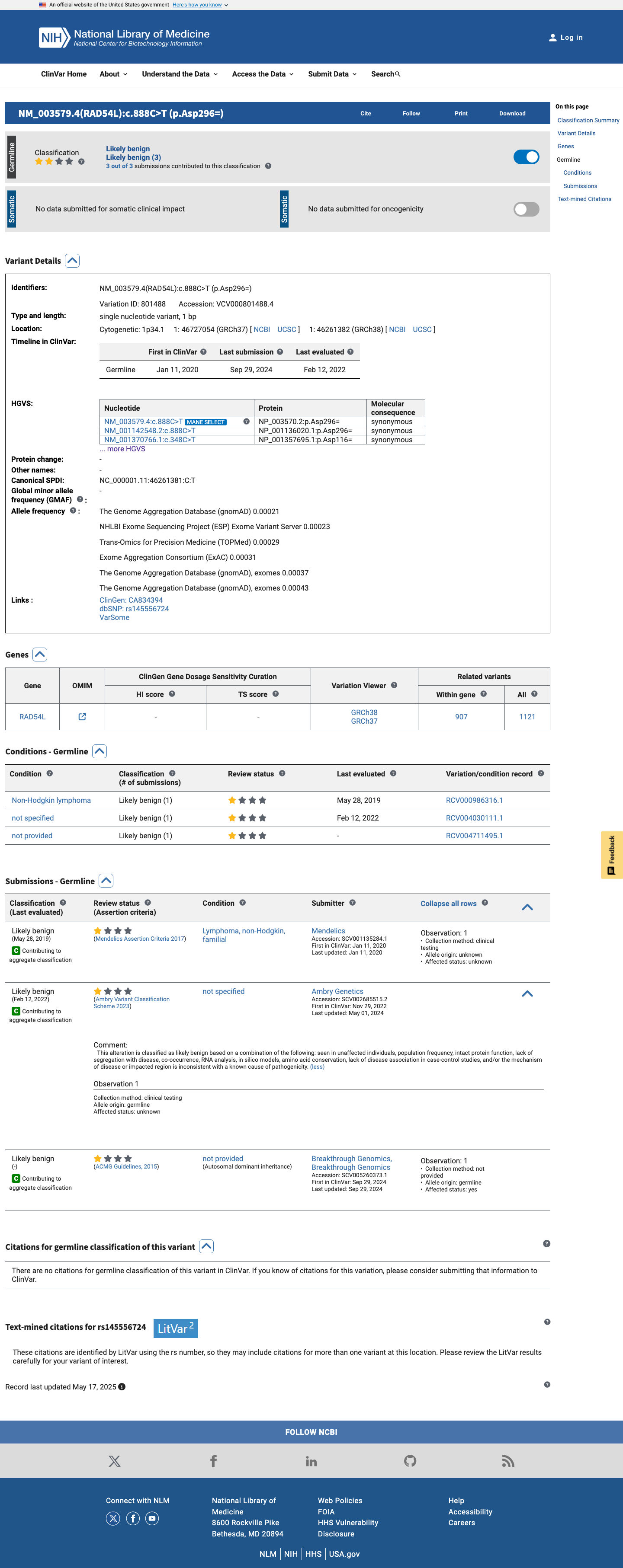
ClinVar
This variant has been reported in ClinVar as Likely benign (2 clinical laboratories). (ClinVarID = 801488)
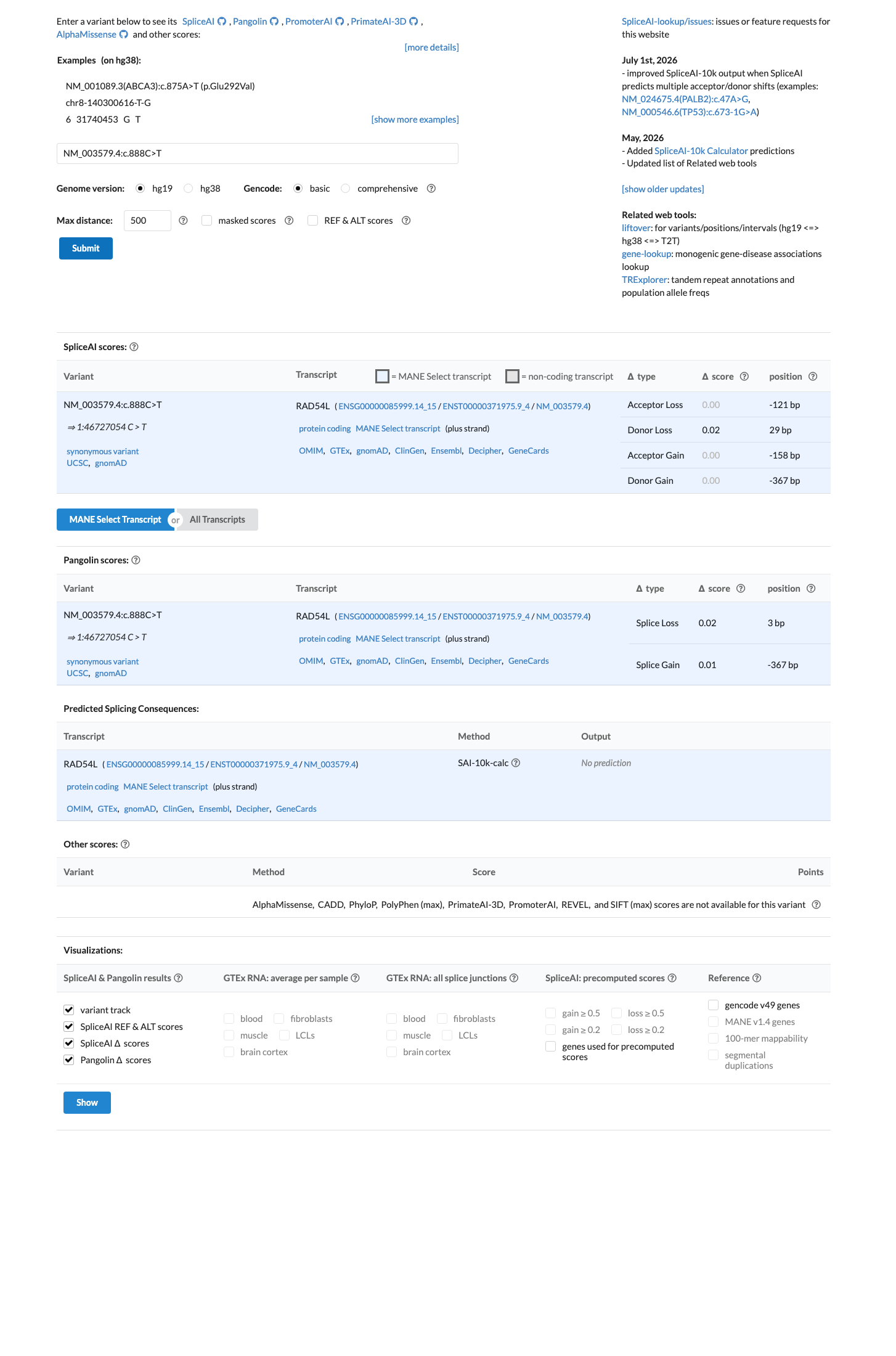
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR