PM2 at supporting: NM_005247.2:c.121G>T (p.Gly41Trp) is present at extremely low frequency in gnomAD v4.1 (AF=0.00195%, 29/1,489,482 alleles, 0 homozygotes, grpmax FAF=1.52e-05) and is absent from gnomAD v2.1 and gnomAD-Canada.1 BP4 at supporting_benign: Multiple in silico algorithms predict a benign effect (REVEL 0.432, BayesDel -0.07, SpliceAI max delta 0.00) with no evidence of splicing impact or hotspot localization.2 PVS1 is not applicable as this is a missense variant, not a null variant (nonsense, frameshift, or canonical splice site).3 No pathogenic or likely pathogenic classification from ClinVar expert panels is available (variant is absent from ClinVar); PS5 and PP5 are not met. PS1 and PM5 are not met as no pathogenic comparator variants exist at residue Gly41.4 No variant-specific functional studies, case-control data, segregation data, de novo reports, or phenotype data are available; PS3, PS4, PS2, PM6, PP1, PP4, BS2, BS4, BP2, and BP5 remain not assessed.5 BA1 and BS1 are not met as the population frequency (AF=0.00195%) is well below both the 1% and 0.3% thresholds respectively.6 Final classification: Variant of Uncertain Significance (VUS). One supporting pathogenic criterion (PM2) and one supporting benign criterion (BP4) are met, which are offsetting. No other criteria are met. Per generic ACMG/AMP 2015 combination rules, this does not reach the threshold for Likely Pathogenic, Likely Benign, Pathogenic, or Benign.7
FGF3
Final classification
VUS
FGF3 c.121G>T · p.Gly41Trp
FGF3
PM2 at supporting: NM_005247.2:c.121G>T (p.Gly41Trp) is present at extremely low frequency in gnomAD v4.1 (AF=0.00195%, 29/1,489,482 alleles, 0 homozygotes, grpmax FAF=1.52e-05) and is absent from gnomAD v2.1 and gnomAD-Canada.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
FGF3 c.121G>T
PM2 + BP4
→
VUS
Gene diagram
· NM_005247.2 · variants mapped to exon structure
FGF3
NM_005247.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 21 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in gnomAD v4.1 (AF=0.00195%, 29/1,489,482 alleles, 0 homozygotes, grpmax FAF=1.52e-05) and is absent from gnomAD v2.1 and gnomAD-Canada. The frequency is well below the 0.1% PM2 threshold.
gnomAD v4.1: AF=0.00195% (29 alleles)grpmax FAF=1.52e-05. Absent from gnomAD v2.1 and gnomAD-Canada.
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no impact on the gene product. REVEL score is 0.432 (below deleterious threshold), BayesDel score is -0.07 (predicts benign), and SpliceAI max delta is 0.00 (no predicted splicing impact). The variant is not in a mutational hotspot.
REVEL: 0.432 (below 0.5). BayesDel: -0.07 (benign). SpliceAI: max delta 0.00. Hotspots: not significant.
Assessed · not applied
Pathogenic
PS1
No pathogenic or likely pathogenic variant at the same amino acid position (Gly41) with a different nucleotide change has been identified in ClinVar or the literature to satisfy PS1.
PS2
No de novo data (paternity not confirmed or unconfirmed) are available for this variant in any database or literature source.
PS3
No variant-specific functional data have been identified.
PS4
No case-control data or statistical enrichment analysis is available for this variant in affected individuals versus controls.
PM1
This variant (p.Gly41Trp) does not lie in a statistically significant hotspot per cancerhotspots.org, and no literature was identified characterizing position 41 as a critical functional domain in FGF3.
PM5
No pathogenic or likely pathogenic missense variant at the same amino acid position (Gly41) with a different amino acid change was identified in ClinVar.
PM6
No de novo data (paternity not confirmed or unconfirmed) are available for this variant.
PP1
No segregation data are available for this variant.
PP2
No gene-level missense constraint metrics (e.g., gnomAD missense Z-score) or evidence that missense variants are a common disease mechanism for FGF3 are available to assess PP2.
PP3
In silico predictions do not support a deleterious effect.
PP4
No patient phenotype data are available to assess whether the variant carrier's clinical presentation is specific for FGF3-related disease.
PP5
This variant is absent from ClinVar; no reputable source or expert panel has classified it as pathogenic.
Benign
BA1
The variant's maximum allele frequency in gnomAD v4.1 is 0.00195% (AF=1.95e-05) with grpmax FAF of 1.52e-05, well below the BA1 threshold of >1%.
BS1
The variant's allele frequency in gnomAD v4.1 is 0.00195%, below the BS1 threshold of >0.3%.
BS2
No data are available regarding observation of this variant in healthy adults with full penetrance expected at an early age.
BS3
No well-established in vitro or in vivo functional studies showing no damaging effect on protein function or splicing have been identified for this variant.
BS4
No segregation data are available to assess lack of cosegregation with disease.
BP1
While FGF3 loss-of-function is supported as a germline disease mechanism, there is no evidence that only truncating variants cause disease in FGF3.
BP2
No data are available regarding observation of this variant in trans with a pathogenic variant in FGF3.
BP5
No data are available regarding an alternate molecular basis for disease in the proband.
BP6
This variant is absent from ClinVar; no reputable source has classified it as benign or likely benign.
N/A · 5
PVS1 · PM3 · PM4 · BP3 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
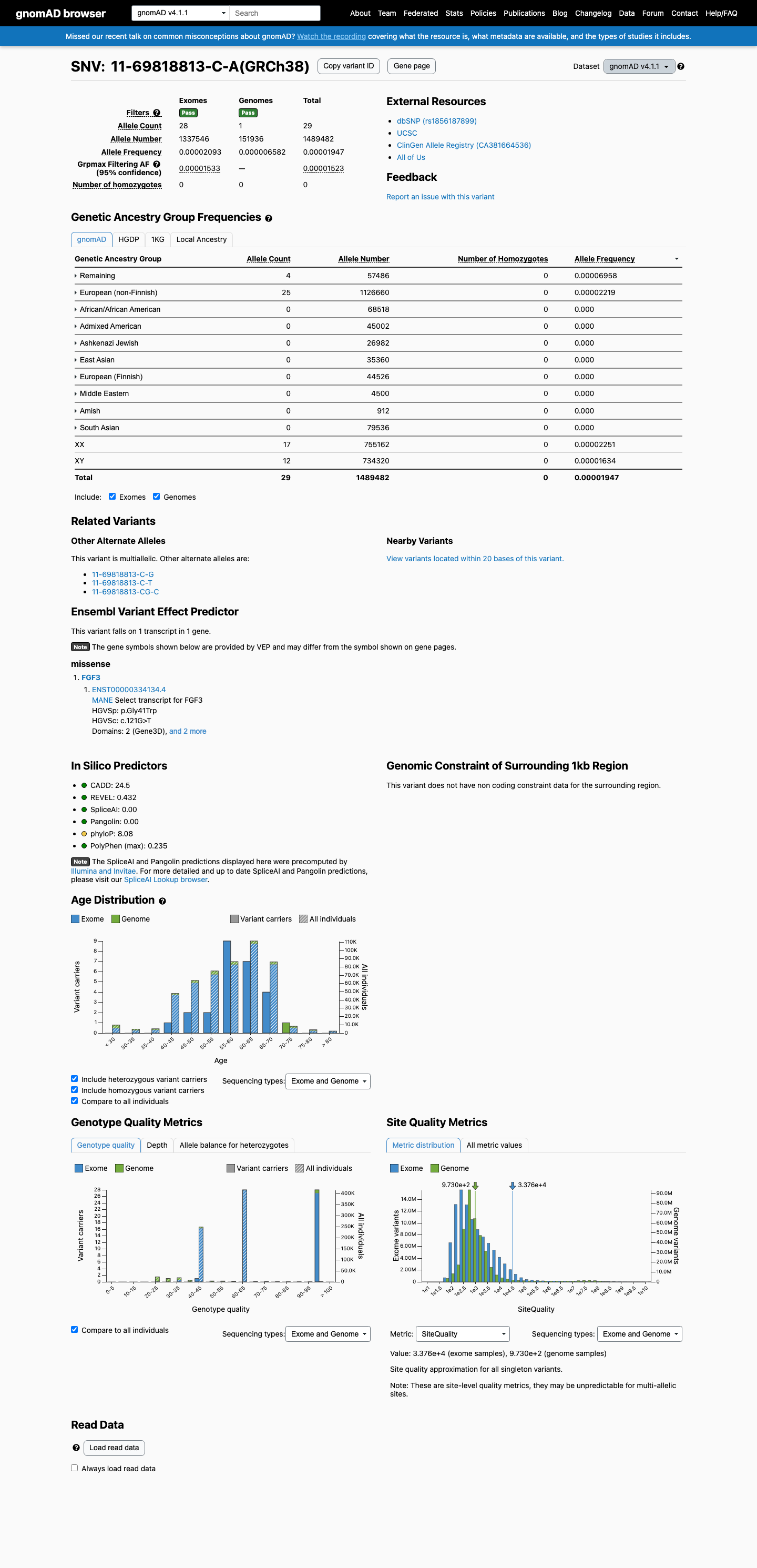
This variant is present in gnomAD v4.1 (AF= 1.94699e-05; MAF= 0.00195%, 29/1489482 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 6.95822e-05; MAF= 0.00696%, 4/57486 alleles, homozygotes = 0); grpmax FAF= 1.523e-05.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0019%
· 29 / 1,489,482
0 hom · FAF 0.0015%
0 hom · FAF 0.0015%
Remaining individuals 4 / 57,486 |
0.007% |
European (non-Finnish) 25 / 1,126,660 |
0.0022% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

In silico
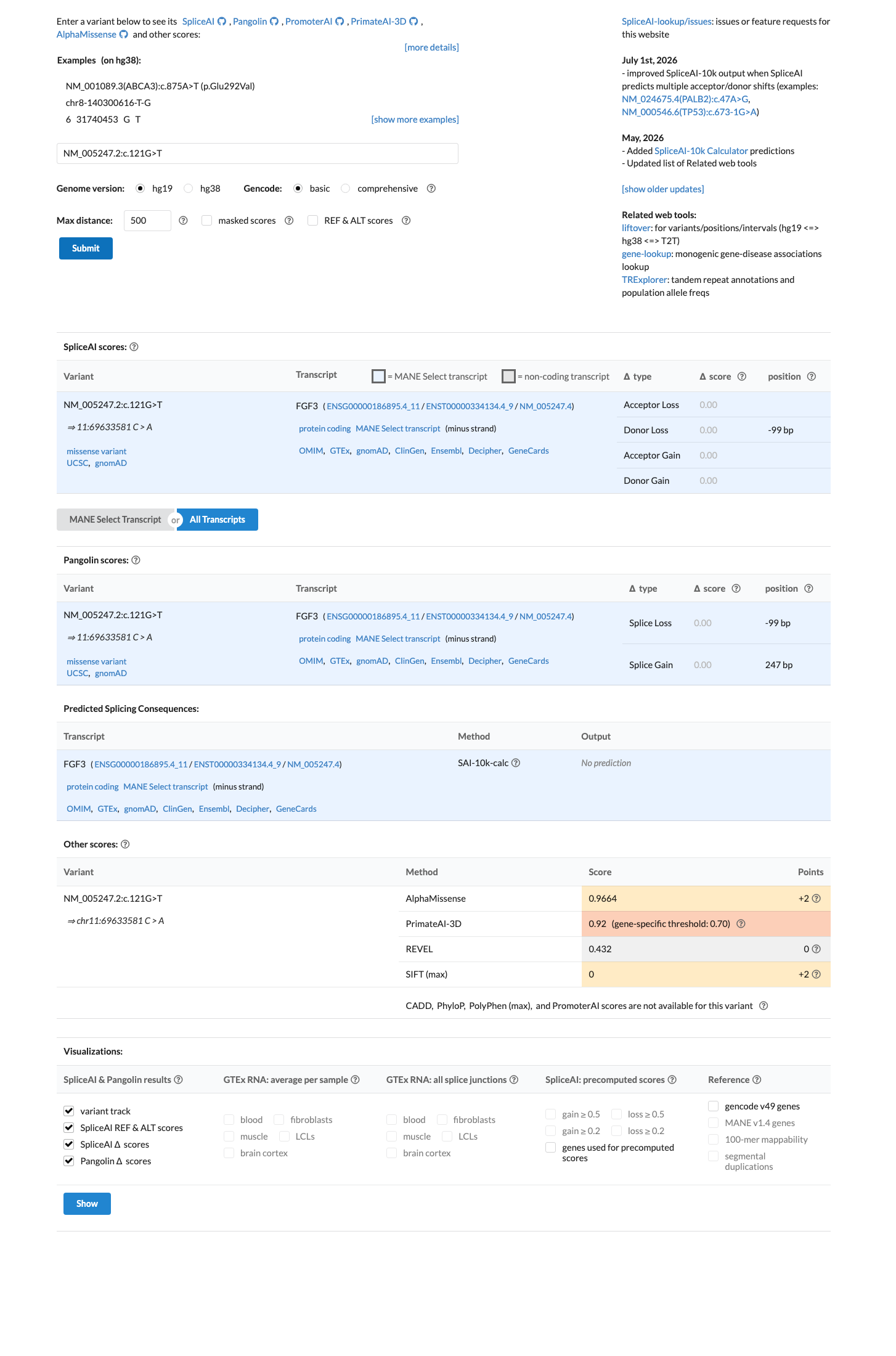
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.432. BayesDel score = -0.0702942.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. FGF3, a fibroblast growth factor, is altered by amplification in various cancer types.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links