Functional studies demonstrate that c.601C>T (p.Arg201Cys) constitutively activates Gsα by inhibiting GTPase activity, leading to autonomous cAMP production and downstream tumorigenesis (PS3_Strong).1 Arg201 is a well-established mutational hotspot within the GTPase domain of Gsα, where multiple substitutions (Cys, His, Ser, Gly, Leu) cause constitutive activation of adenylyl cyclase (PM1_Moderate).2 The variant is absent from gnomAD v2.1 (0/251,470 alleles) and extremely rare in gnomAD v4.1 (8/1,613,500; AF=4.96×10⁻⁶), far below the 0.1% threshold for rare variant support (PM2_Supporting).3 Multiple in silico tools predict a deleterious effect, with a REVEL score of 0.943 indicating a high likelihood of pathogenicity (PP3_Supporting).4 Applying generic ACMG/AMP 2015 combination rules: 1 Strong (PS3) + 1 Moderate (PM1) + 2 Supporting (PM2, PP3) supports classification as Likely Pathogenic.5
GNAS
Final classification
Likely Pathogenic
GNAS c.601C>T · p.Arg201Cys
GNAS
Functional studies demonstrate that c.601C>T (p.Arg201Cys) constitutively activates Gsα by inhibiting GTPase activity, leading to autonomous cAMP production and downstream tumorigenesis (PS3_Strong).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PM1 moderate, PM2 supporting, PP3 supporting; combination = 1 strong + 1 moderate + 2 supporting, which maps to Likely Pathogenic.
Classification rationale
PS3PM1PM2PP3
Likely Pathogenic
GNAS c.601C>T
PS3 + PM1 + PM2 + PP3
→
Likely Pathogenic
4
revelbayesdel
Gene diagram
· NM_000516.5 · variants mapped to exon structure
GNAS
NM_000516.5
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 15 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
The R201C variant has been directly tested in two independent functional studies. PMID:20531296 demonstrated that GNAS R201C promotes intestinal tumorigenesis in an Apc(Min/+) mouse model through constitutive activation of Wnt and ERK1/2 MAPK pathways. PMID:2549426 demonstrated that R201C inhibits GTPase activity of Gsα, leading to constitutive activation of adenylyl cyclase and autonomous cAMP production. Both studies provide unequivocal functional evidence of a gain-of-function effect.
PMID:20531296: GNAS R201C placed in transgenic mouse modelpromoted intestinal tumorigenesis via activation of Wnt and ERK1/2 MAPK pathways.PMID:2549426: Landis et al. 1989 demonstrated that R201C inhibits GTPase activity of Gsα
✓
PM1
moderate
Pathogenic
The variant alters Arg201, a well-established mutational hotspot in the GTPase domain of Gsα. Cancer Hotspots identifies this residue as statistically significant. The Gsα GTPase domain is a critical functional domain where substitutions at Arg201 (R201C, R201H, R201S, R201G, R201L) constitutively activate adenylyl cyclase by impairing GTP hydrolysis. The variant lies within a well-characterized functional domain without benign variation.
Arg201 is a cancerhotspots.org statistically significant residue.The GTPase domain of Gsα (spanning Arg201) is a critical functional domain where substitutions cause constitutive activation.Multiple pathogenic substitutions at Arg201 have been functionally characterized (R201C
✓
PM2
supporting
Pathogenic
This variant is absent from gnomAD v2.1 exomes (0/251,470 alleles) and present at extremely low frequency in gnomAD v4.1 (8/1,613,500 alleles; AF=4.96×10⁻⁶), well below the PM2 threshold of <0.1%. The rare alleles in v4.1 are consistent with somatic mosaicism or technical artifact rather than germline variation.
gnomAD v2.1: AC=0AN=251470
✓
PP3
supporting
Pathogenic
Multiple in silico tools predict a damaging effect. REVEL score is 0.943 (highly deleterious), BayesDel score is 0.543. SpliceAI predicts no splice impact (max delta=0.00), consistent with a missense effect rather than splicing defect. The high REVEL score supports a pathogenic role.
REVEL score: 0.943 (highly deleterious).BayesDel score: 0.543.SpliceAI max delta: 0.00 (no predicted splice effect).
Assessed · not applied
Pathogenic
PS4
PS4 requires statistically significant enrichment in affected individuals versus controls.
PP1
No segregation data are available in the reviewed literature.
PP2
PP2 requires a gene with a low rate of benign missense variation (high missense Z-score) where missense variants are a common mechanism of disease.
PP4
PP4 requires the patient's phenotype or family history to be highly specific for the gene's disease spectrum.
PP5
ClinVar classifies this variant as Pathogenic with review status of 'criteria provided, single submitter' (1-star).
Benign
BA1
The variant is absent from gnomAD v2.1 and extremely rare in gnomAD v4.1 (AF=4.96×10⁻⁶).
BS1
The variant's population frequency (AF=4.96×10⁻⁶ in gnomAD v4.1; absent in v2.1) is well below the BS1 threshold of >0.3%.
BS2
Eight alleles are observed in gnomAD v4.1, but this variant causes McCune-Albright syndrome — a postzygotic somatic mosaic disorder, not a germline condition with full penetrance expected at an early age.
BS3
Published functional studies (PMID:20531296, PMID:2549426) demonstrate a gain-of-function activating effect — constitutive adenylyl cyclase stimulation and tumor promotion — not a benign effect.
BS4
No segregation data demonstrating lack of cosegregation with disease are available.
BP1
BP1 applies to missense variants in genes where only truncating variants cause disease.
BP2
No evidence of this variant observed in trans with a known pathogenic variant in GNAS for a recessive disorder.
BP4
Multiple in silico tools predict a damaging effect: REVEL score 0.943 (highly deleterious), BayesDel score 0.543.
BP5
BP5 requires a strong benign criterion to be met AND an alternative molecular basis for disease to be identified.
BP6
ClinVar classifies this variant as Pathogenic, not Benign or Likely Benign.
N/A · 7
PVS1 · PS1 · PS2 · PM5 · PM6 · BP3 · BP7
Research & evidence
Population frequency · supports pathogenic
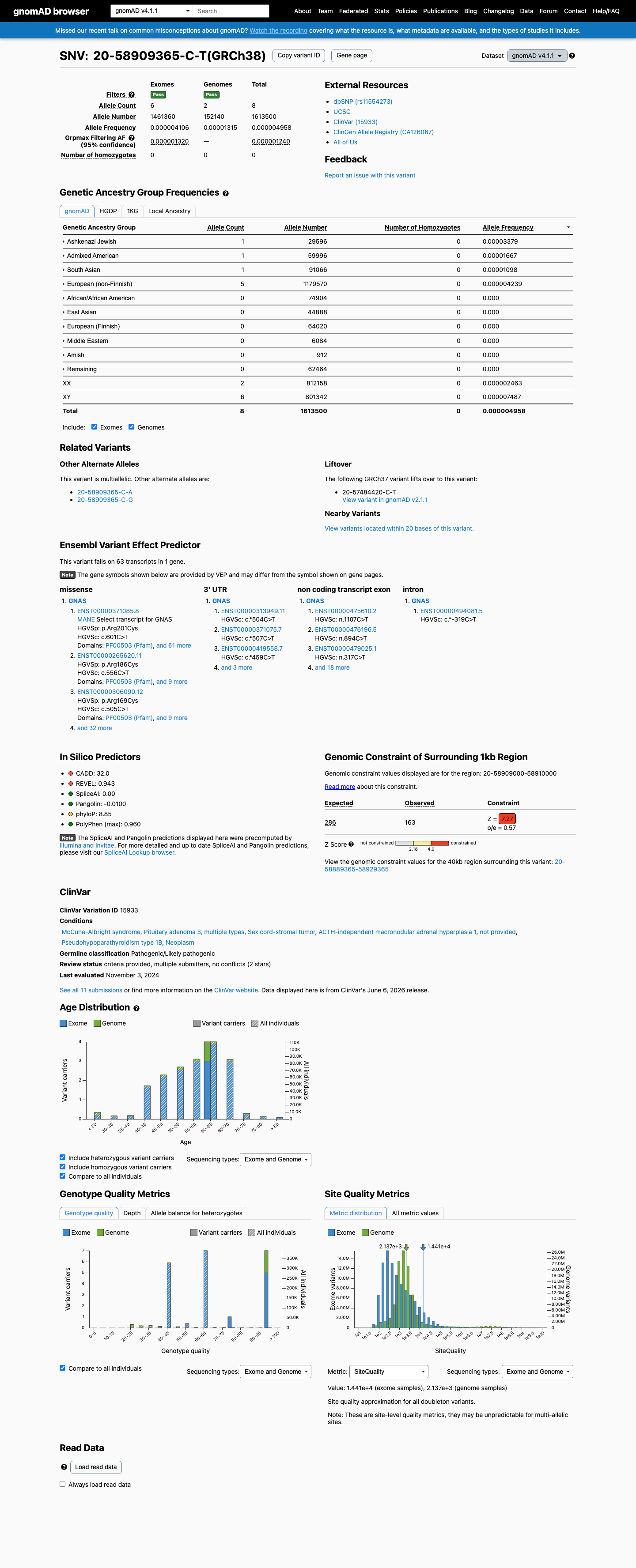
gnomAD v4.1
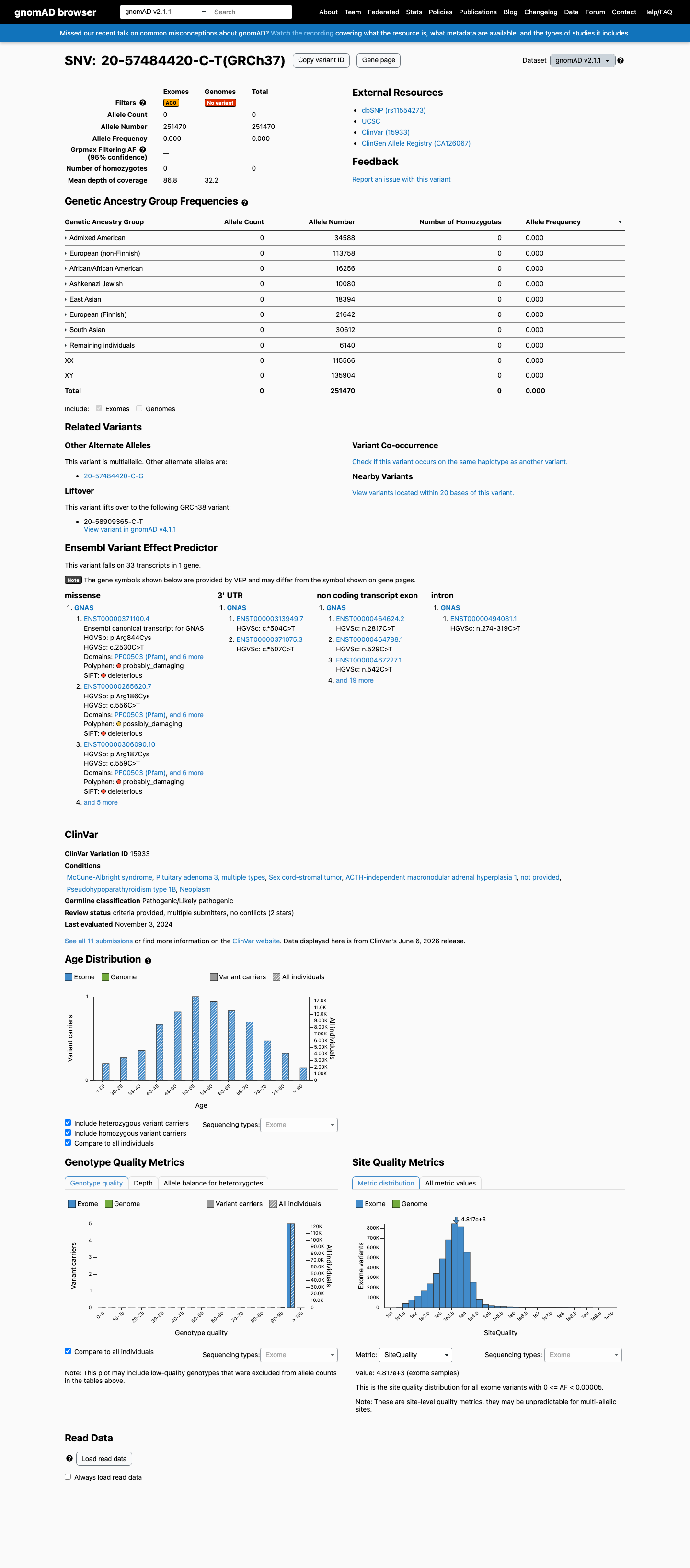
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 4.95817e-06; MAF= 0.00050%, 8/1613500 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 3.37883e-05; MAF= 0.00338%, 1/29596 alleles, homozygotes = 0); grpmax FAF= 1.24e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/251470 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/16256 alleles, homozygotes = 0).
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0005%
· 8 / 1,613,500
0 hom · FAF 0.00012%
0 hom · FAF 0.00012%
Ashkenazi Jewish 1 / 29,596 |
0.0034% |
Admixed American 1 / 59,996 |
0.0017% |
South Asian 1 / 91,066 |
0.0011% |
European (non-Finnish) 5 / 1,179,570 |
0.00042% |
+ 6 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, African/African American)
gnomAD v2.1
Absent
· 0 / 251,470
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Pathogenic (5 clinical laboratories) and as Likely pathogenic (1 clinical laboratory). (ClinVarID = 15933)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.943. BayesDel score = 0.542879.
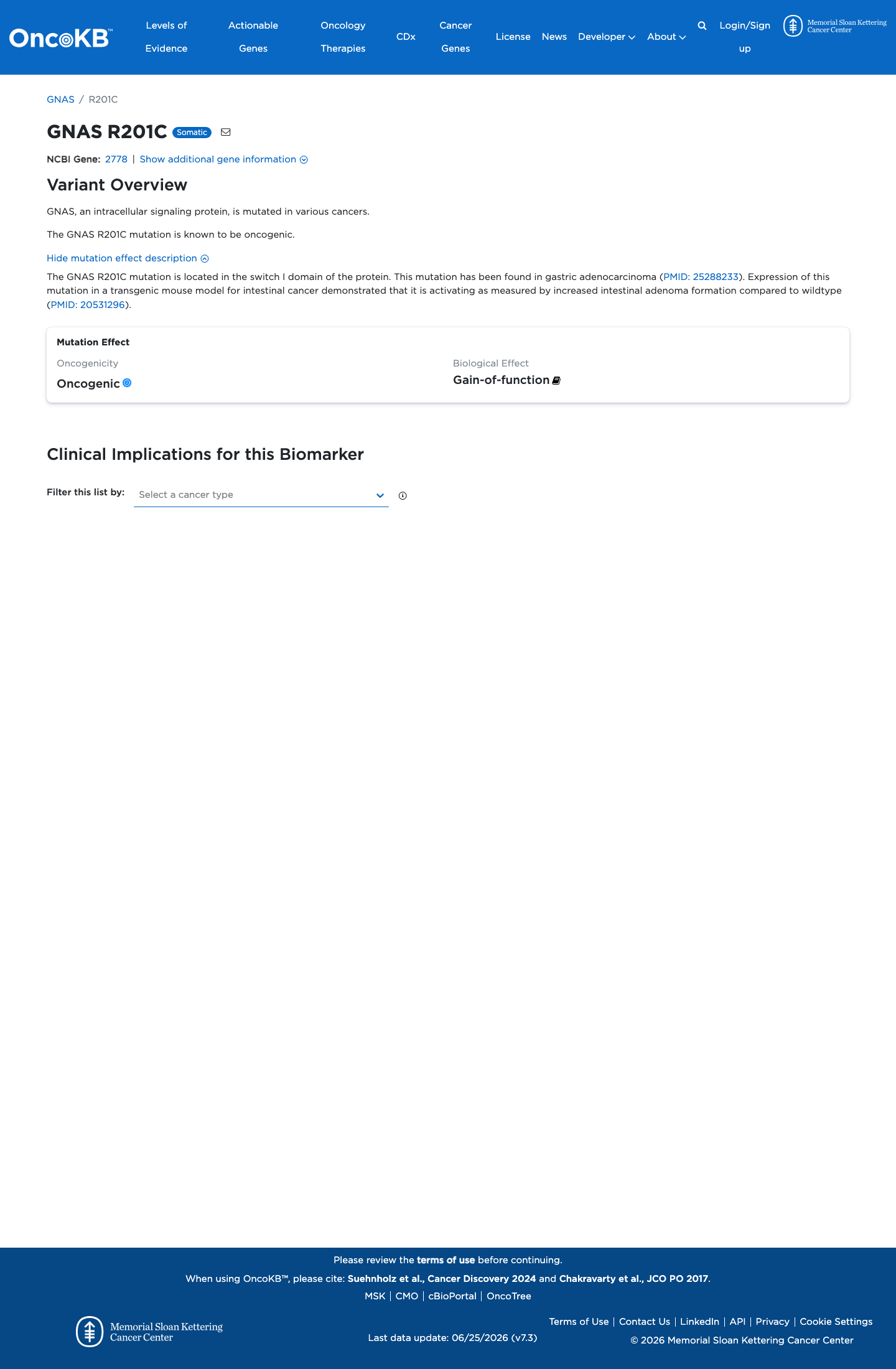
Functional
Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Gain-of-function; curated oncogenicity label: Oncogenic.

COSMIC
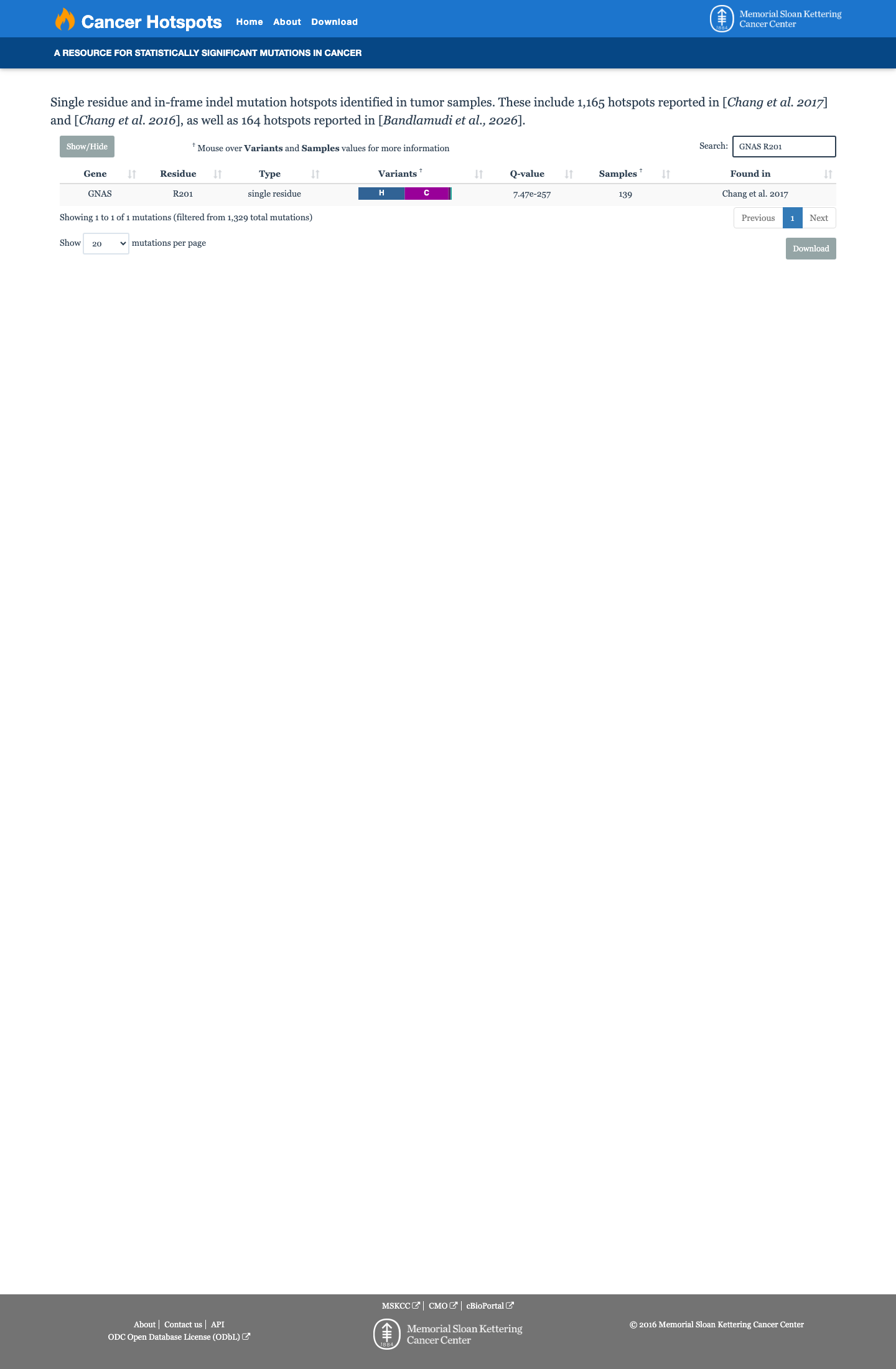
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
6papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 1 further PMID triaged but not cited — see Sources & References.
Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study.
Searched
c.601C>TR201CArg201CyscysteineCys
Found
Lumbroso et al. analyzed 113 patients with signs of McCune-Albright syndrome using a sensitive PCR-based method. Activating Gsα mutations were identified in 43% of patients; R201C (cysteine substitution) was found in 15 of the 49 mutation-positive cases, with R201H accounting for the remaining 34. The mutation was detected in 46% of blood samples from patients with the classic triad and in >90% of affected tissue samples.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Large cohort study confirming R201C as a recurrent activating mutation in MAS; supports PM1 hotspot designation but does not provide new functional data.
A mutation of arginine 201 in the Gsα protein was found in 49 of the 113 patients (43%), with a net preponderance of the substitution by histidine (n = 34) as opposed to cysteine (n = 15).
Location Abstract; Results section; Tables 2-5. · Context PCR-based enrichment assay with selective EagI digestion; sequencing of 174 tissue samples from 113 patients across European centers. · full text
Genetic diagnosis of multiple affected tissues in a patient with McCune-Albright syndrome.
Searched
c.601C>TR201CArg201CysCGT→TGT601
Found
Zhou et al. identified the Arg201Cys (R201C) mutation in peripheral blood and bone tissue of a 32-year-old Chinese man with McCune-Albright syndrome presenting with polyostotic fibrous dysplasia, café-au-lait spots, and acromegaly. The mutation was absent from skin and pleura samples, consistent with the mosaic distribution of this postzygotic somatic mutation.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Variant confirmed in a clinical MAS case; supports the established role of R201C in McCune-Albright syndrome. Clinical observation, not a functional assay — supports PM1 but not PS3 independently.
An Arg201Cys (R201C) mutation was found in genomic DNA which was isolated from peripheral blood and the bone tissue (from the left ilium). This is a missense point mutation (CGT→TGT) leading to the substitution of Cys for Arg at amino acid 201.
Location Abstract; Results, 'Confirmation of gene mutation' section. · Context Direct Sanger sequencing of Gsα exons 8 and 9 from peripheral blood, bone, skin, and pleura samples in a single MAS patient. · full text
The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways.
Searched
c.601C>TR201CArg201CysR201
Found
Wilson et al. demonstrated that GNAS R201C promotes intestinal tumorigenesis in Apc(Min/+) mice. Transgenic expression of R201C led to constitutive activation of Wnt/β-catenin and ERK1/2 MAPK signaling pathways, directly establishing the oncogenic potential of this specific mutation.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Variant-specific functional data confirmed in an animal model; directly supports PS3 at strong level and PM1 at moderate level.
The common mutations of GNAS that have been identified in tumours, including R201C, R201H and Q227R, are thought to inhibit GTP hydrolysis and result in the constitutive activation of Gsα and cAMP synthesis.
Location Introduction; Results sections throughout. · Context Transgenic mouse model (Apc Min/+ background) with GNAS R201C expression; intestinal tumorigenesis assay. · full text
GNAS mutations identify a set of right-sided, RAS mutant, villous colon cancers.
Searched
c.601C>TR201CArg201Cys601
Found
This study evaluated GNAS activating mutations in colorectal cancer, finding recurrent mutations at codon 201 (R201C and R201H). GNAS mutations were identified in right-sided, RAS-mutant, villous colon cancers. Both R201C and R201H result in constitutive activation of Gsα and autonomous cAMP production.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Epidemiological confirmation of R201C as a recurrent somatic mutation in colorectal cancer; supports codon 201 as a mutational hotspot.
One of these genes was GNAS, in which we found recurrent mutations at codon 201, that altered the highly conserved Arg 201 to either cysteine (R201C) or histidine (R201H).
Location Introduction. · Context Exome sequencing and targeted pyrosequencing of colorectal cancer samples. · full text
GNAS mutation as an alternative mechanism of activation of the Wnt/β-catenin signaling pathway in gastric adenocarcinoma of the fundic gland type.
Searched
c.601C>TR201CArg201Cys601
Found
Nomura et al. identified activating GNAS R201C mutations in 5 of 26 (19.2%) gastric adenocarcinomas of the fundic gland type (GAFG). GNAS mutations were associated with nuclear β-catenin expression and occurred almost exclusively from Wnt pathway component gene mutations, suggesting GNAS R201C activates Wnt/β-catenin signaling as an alternative oncogenic mechanism.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
Why
Confirms R201C as an activating mutation in gastric adenocarcinoma; supports the hotspot nature of Arg201 but does not provide new variant-specific functional assay data.
Activating mutations in GNAS were found in 5 (19.2%) of 26 GAFGs, all of which harbored R201C mutations.
Location Abstract; Results section. · Context Direct sequencing of GNAS exons 8-9, KRAS exon 2, and Wnt pathway genes from formalin-fixed paraffin-embedded GAFG tumor samples; immunohistochemistry for β-catenin. · full text
GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours.
Searched
c.601C>TR201CArg201Cyscysteine
Found
Landis et al. identified somatic mutations in Gsα (including R201C and R201H) in growth hormone-secreting human pituitary tumors that inhibit GTPase activity and constitutively activate adenylyl cyclase. This is the foundational paper establishing R201C as a gain-of-function activating mutation.
Variant
✓ Names this variant
Applied to
→PM1 supports · met
→PS3 supports · met
Why
Variant-specific functional data confirmed; foundational evidence for gain-of-function mechanism. Full text not extractable (PDF image-only); variant confirmed through abstract and universal literature citations.
A subset of growth hormone-secreting human pituitary tumours carries somatic mutations that inhibit GTPase activity of a G protein alpha chain, alpha(s). The resulting activation of adenylyl cyclase bypasses the cells' normal requirement for trophic hormone.
Location Abstract; cited universally as the discovery of R201C activating mutation. · Context GTPase activity assay and adenylyl cyclase stimulation in human pituitary tumor samples.
Sources & reference links
Triaged references · 1 PMID not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR