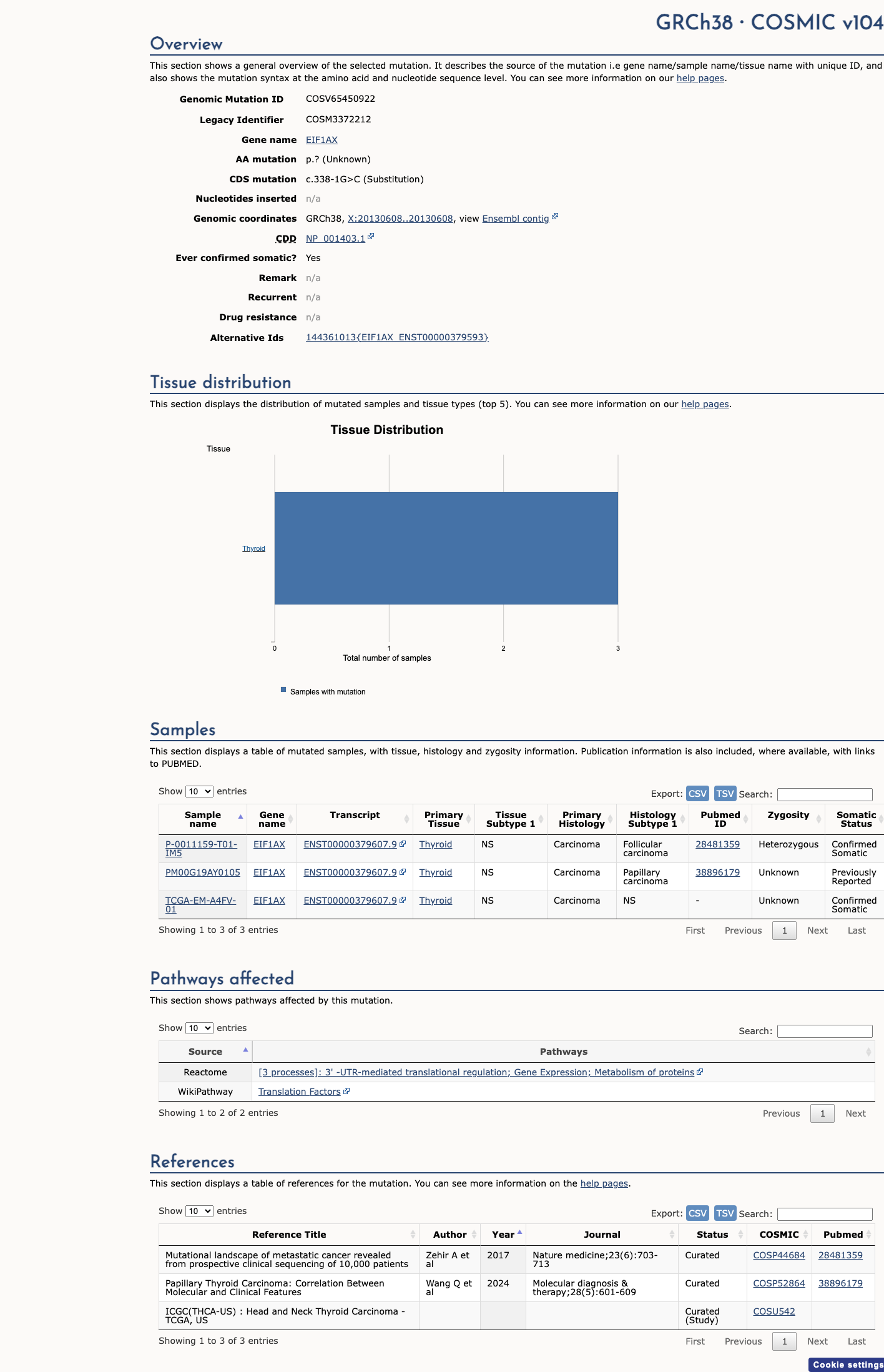
NM_001412.4:c.338-1G>C is a canonical ±1 splice acceptor variant in intron 5 of EIF1AX, a gene in which loss of function is supported as a germline disease mechanism in uveal melanoma predisposition. Under ClinGen SVI PVS1 recommendations (PMC6185798), this qualifies for PVS1 at strong strength.1 The variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada population databases, satisfying PM2 at moderate strength.2 SpliceAI predicts a splice-altering effect (max delta score 0.68, acceptor loss). This computational evidence is already accounted for in PVS1 and is not separately applied as PP3 per PMC6185798 guidance against double-counting splice prediction evidence.3 No de novo data (PS2/PM6), no functional studies (PS3/BS3), no case-control data (PS4), no segregation data (PP1/BS4), and no patient phenotype information (PP4) are available for this variant. These criteria remain unmet. ClinVar variation ID 4484109 has zero submissions, no classification, and no review status. PP5 and BP6 cannot be applied.4 The variant has been observed in 3 somatic tumor samples in COSMIC (COSV65450922). Somatic occurrence does not independently satisfy germline criteria but is consistent with a role in tumorigenesis. The only associated publication (PMID:31275557) describes a pan-cancer splicing mutation repository but does not include NM_001412.4:c.338-1G>C in its dataset. No publication provides variant-specific evidence for any criterion.5 Overall classification: Likely Pathogenic. The combination of PVS1 (strong) and PM2 (moderate) satisfies the generic ACMG/AMP 2015 threshold for Likely Pathogenic (1 strong + 1 moderate).6
EIF1AX
Final classification
Likely Pathogenic
EIF1AX c.338-1G>C · p.?
EIF1AX
NM_001412.4:c.338-1G>C is a canonical ±1 splice acceptor variant in intron 5 of EIF1AX, a gene in which loss of function is supported as a germline disease mechanism in uveal melanoma predisposition. Under ClinGen SVI PVS1 recommendations (PMC6185798), this qualifies for PVS1 at strong strength.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PM2 moderate; combination = 1 strong + 1 moderate, which maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
EIF1AX c.338-1G>C
PVS1 + PM2
→
Likely Pathogenic
1
pvs1_generic_framework ↗pvs1_gene_contextpvs1_variant_assessment
6
generic_acmg_combination_rules
Gene diagram
· NM_001412.4 · variants mapped to exon structure
EIF1AX
NM_001412.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
review
Pathogenic
NM_001412.4:c.338-1G>C is a canonical ±1 splice acceptor variant affecting intron 5 of EIF1AX. Under ClinGen SVI PVS1 recommendations (PMC6185798), canonical splice variants in genes where loss of function is an established disease mechanism are assigned PVS1. EIF1AX germline loss of function is supported as a disease mechanism in uveal melanoma predisposition based on targeted literature review. The MANE Select transcript NM_001412.4 is biologically relevant. No evidence of nonsense-mediated decay escape, alternative splicing of the affected exon, or population LoF enrichment was identified. PVS1 is applied at strong strength given the limited but positive germline LoF evidence base for this gene.
Canonical ±12 splice acceptor variant (c.338-1G>Cintron 5) under PMC6185798
✓
PM2
moderate
Pathogenic
NM_001412.4:c.338-1G>C is absent from gnomAD v2.1 (exomes), gnomAD v4.1 (exomes/genomes), and gnomAD-Canada v1.0. Under generic ACMG/AMP rules, absence from large population databases supports PM2 at moderate strength (allele frequency < 0.1%).
Absent from gnomAD v2.1Absent from gnomAD v4.1Absent from gnomAD-Canada v1.0
Assessed · not applied
Pathogenic
PS2
No de novo data are available for this variant.
PS3
No variant-specific functional data are available for NM_001412.4:c.338-1G>C.
PS4
No case-control or prevalence data are available.
PM1
This variant is a canonical splice site (c.338-1G>C, intron 5 acceptor) and does not fall within a characterized functional domain or mutational hotspot for missense alterations.
PM6
No de novo data are available for NM_001412.4:c.338-1G>C.
PP1
No segregation data are available for this variant.
PP4
No patient phenotype information is available for this variant.
PP5
ClinVar variation ID 4484109 has zero submissions, no classification, and no review status (no star rating).
Benign
BA1
BA1 requires an allele frequency > 1% in population databases.
BS1
BS1 requires an allele frequency > 0.3% in population databases (non-VCEP generic threshold).
BS2
No evidence is available regarding observation of this variant in healthy adults.
BS3
No functional studies demonstrating a neutral effect of NM_001412.4:c.338-1G>C have been identified.
BS4
No segregation data in affected families are available.
BP2
No evidence of observation in trans with a pathogenic variant is available.
BP4
BP4 requires multiple lines of computational evidence to suggest no impact on the gene product.
BP5
No evidence is available that this variant has been observed in a case with an alternative molecular basis for disease.
BP6
ClinVar variation ID 4484109 has zero submissions, no classification, and no review status (no star rating).
N/A · 6
PS1 · PM5 · PP2 · PP3 · BP1 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar but submission details could not be extracted. (ClinVarID = 4484109)
In silico
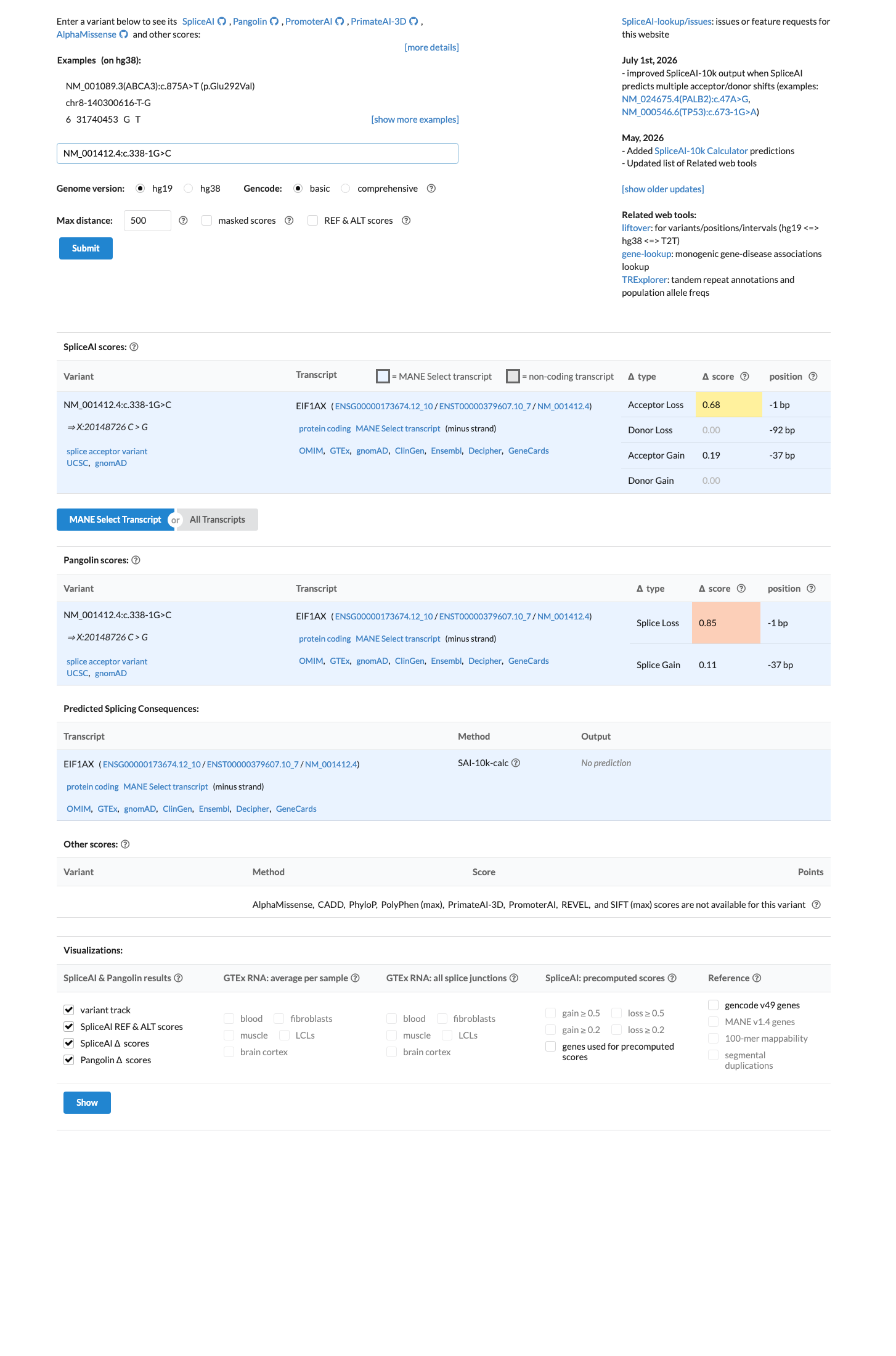
SpliceAI predicts possible splice impact for this variant (max delta score = 0.68). BayesDel score = 0.249774.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV65450922, n = 3 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 1 PMID not cited in assessment