NM_000021.3(PSEN1):c.1234G>A (p.Val412Ile) is extremely rare in population databases, with an allele frequency of 0.00107% in gnomAD v2.1 (3/280,740 alleles) and 0.00143% in gnomAD v4.1 (23/1,613,504 alleles), meeting PM2 at supporting level.1 The variant co-segregates with early-onset frontotemporal dementia in 4 affected individuals across 2 generations in the FUS family (Bernardi et al. 2009, PMID:18314228), meeting PP1 at supporting level. However, incomplete penetrance is noted with 2 cognitively healthy carriers identified at ages 49 and 71 in follow-up (Bernardi et al. 2011, PMID:21297264).2 No variant-specific functional data are available. The in vitro study of 138 PSEN1 pathogenic mutations by Sun et al. (PMID:27930341) did not include p.Val412Ile, as it is currently classified as a variant of uncertain significance.3 In silico predictors are mixed: REVEL score is 0.679, BayesDel is 0.305, and SpliceAI predicts no splicing impact (delta=0.00). The ClinVar submitter LabCorp notes 3 of 5 in silico tools predict a benign effect, precluding application of PP3.4 This variant is classified as Uncertain significance by 3 clinical laboratories in ClinVar (ID: 1318725). No submitter classifies it as pathogenic or likely pathogenic.5 Overall, the evidence for pathogenicity is limited to one supporting criterion (PM2) and one supporting criterion (PP1). With only 2 supporting criteria met and no moderate or strong pathogenic criteria, the variant does not reach the threshold for Likely Pathogenic (which requires 2 supporting + 1 moderate, or variants thereof). The classification remains Uncertain significance.6
PSEN1
Final classification
VUS
PSEN1 c.1234G>A · p.Val412Ile
PSEN1
NM_000021.3(PSEN1):c.1234G>A (p.Val412Ile) is extremely rare in population databases, with an allele frequency of 0.00107% in gnomAD v2.1 (3/280,740 alleles) and 0.00143% in gnomAD v4.1 (23/1,613,504 alleles), meeting PM2 at supporting level.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, PP1 supporting; combination = 2 supporting, which maps to VUS.
Classification rationale
PM2PP1
VUS
PSEN1 c.1234G>A
PM2 + PP1
→
VUS
4
revelbayesdelspliceai ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_000021.3 · variants mapped to exon structure
PSEN1
NM_000021.3
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
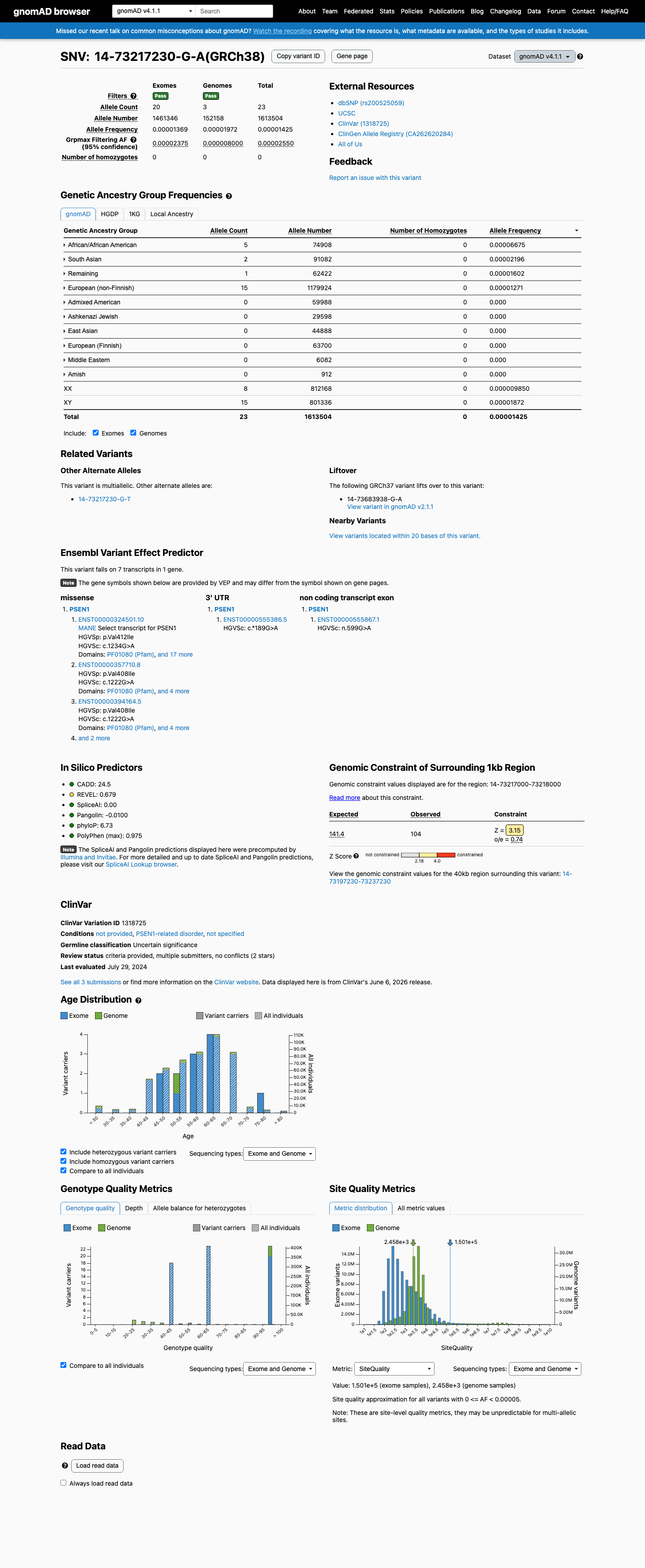
This variant is present in gnomAD v4.1 (AF= 1.42547e-05; MAF= 0.00143%, 23/1613504 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 6.67485e-05; MAF= 0.00667%, 5/74908 alleles, homozygotes = 0); grpmax FAF= 2.55e-05.
v2.1
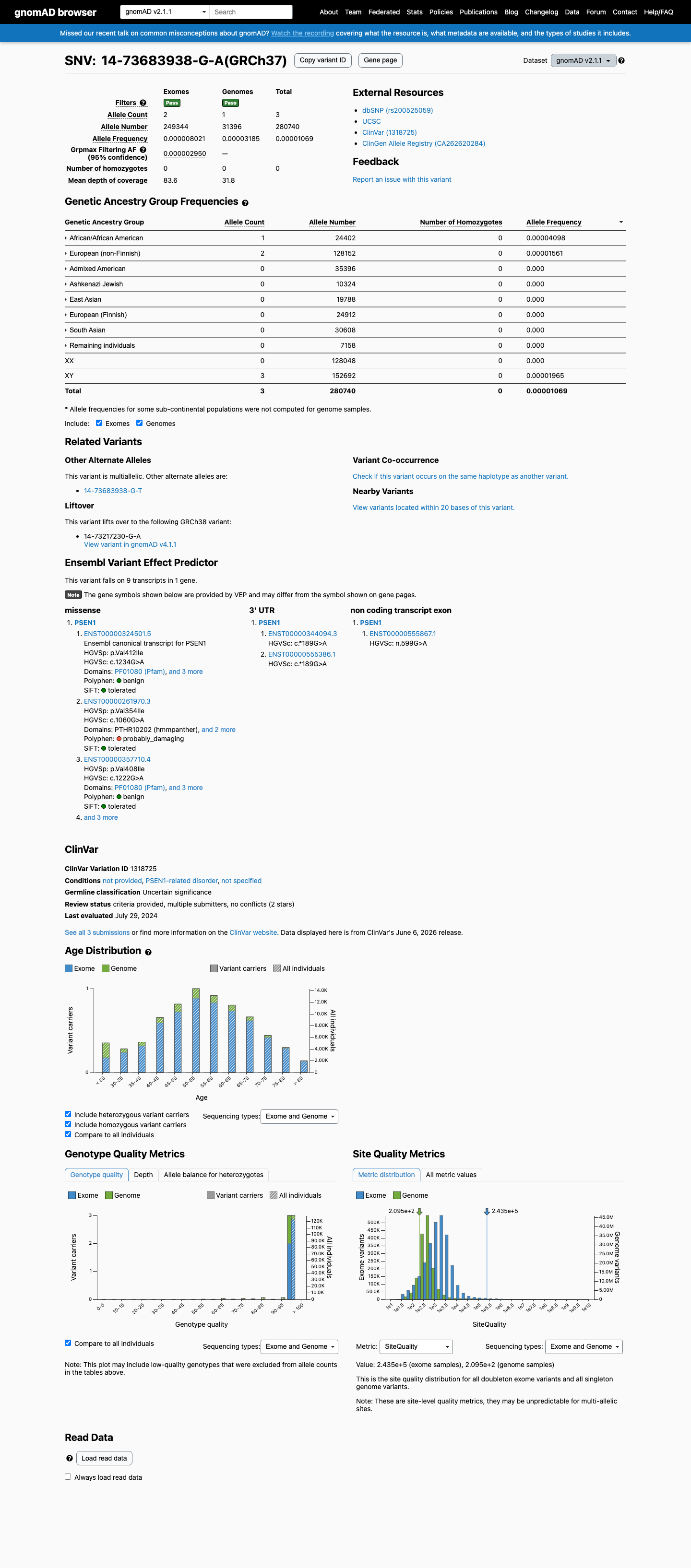
This variant is present in gnomAD v2.1 (AF= 1.0686e-05; MAF= 0.00107%, 3/280740 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 4.09802e-05; MAF= 0.00410%, 1/24402 alleles, homozygotes = 0); grpmax FAF= 2.95e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0014%
· 23 / 1,613,504
0 hom · FAF 0.0026%
0 hom · FAF 0.0026%
African/African American 5 / 74,908 |
0.0067% |
South Asian 2 / 91,082 |
0.0022% |
Remaining individuals 1 / 62,422 |
0.0016% |
European (non-Finnish) 15 / 1,179,924 |
0.0013% |
+ 6 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0011%
· 3 / 280,740
0 hom · FAF 0.0003%
0 hom · FAF 0.0003%
African/African American 1 / 24,402 |
0.0041% |
European (non-Finnish) 2 / 128,152 |
0.0016% |
+ 6 not observed (Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
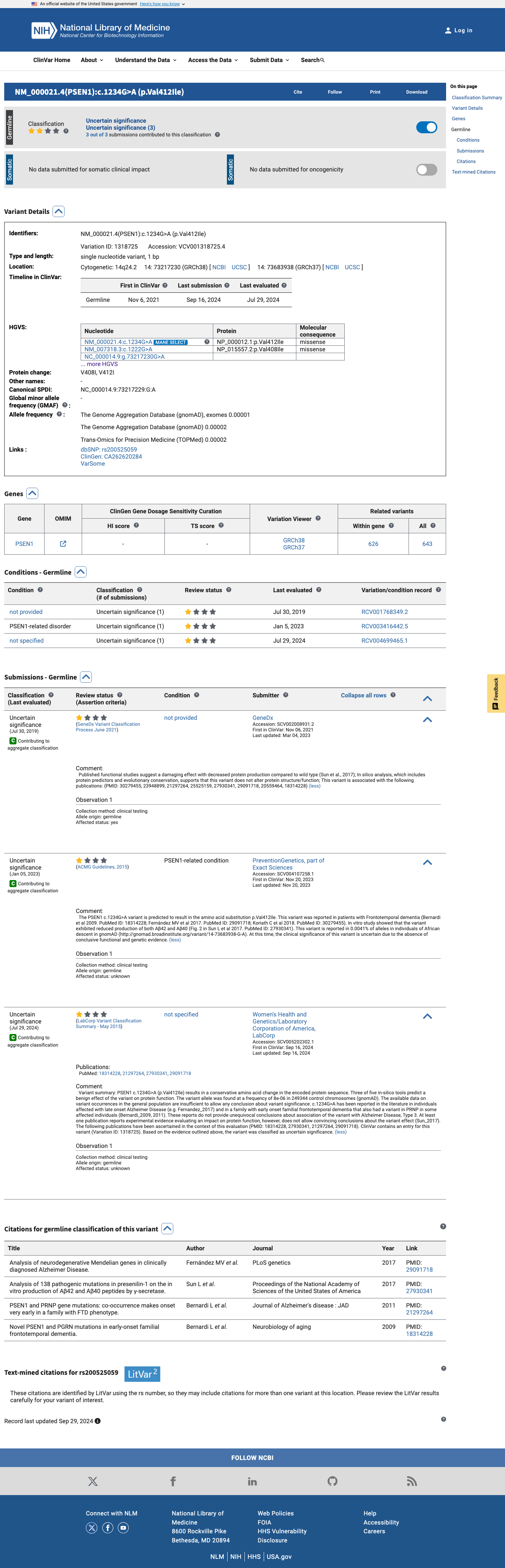
This variant has been reported in ClinVar as Uncertain significance (3 clinical laboratories). (ClinVarID = 1318725)
In silico
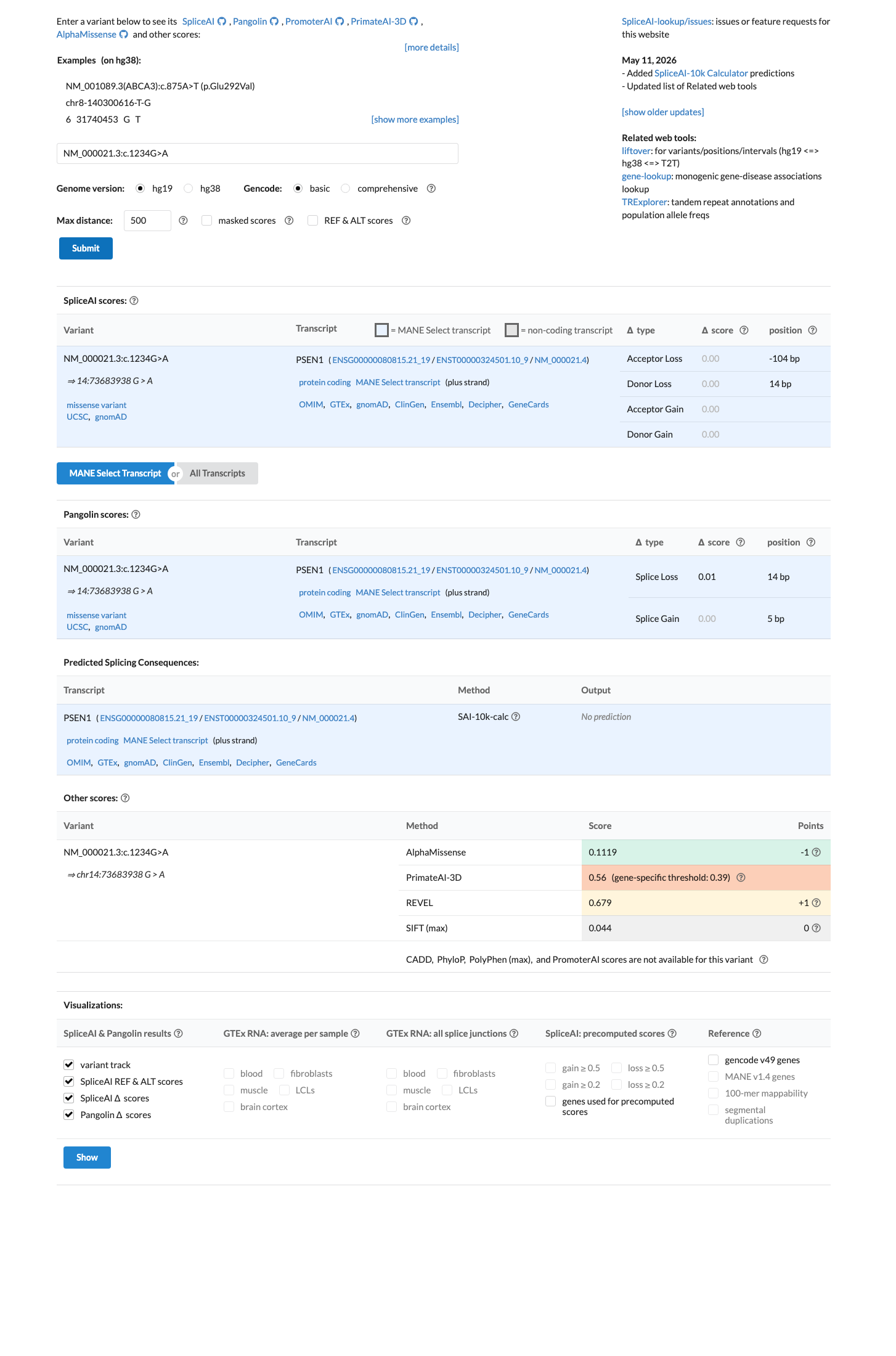
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.679. BayesDel score = 0.305405.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 3 further PMIDs triaged but not cited — see Sources & References.
Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia.
Searched
c.1234G>Ap.Val412IleV412IVal412Ilec.1234
Found
Identified PSEN1 Val412Ile as a novel heterozygous mutation in an autosomal dominant early-onset frontotemporal dementia family (FUS pedigree). The proband, a 38-year-old female, developed progressive mood and personality changes at age 30 with subsequent cognitive decline, mutism, myoclonus, and seizures. Four affected individuals across two generations carried the mutation. Mutation was absent from 100 cognitively healthy controls and 100 previously screened familial Alzheimer disease patients.
Variant
✓ Names this variant — characterised directly
Applied to
→PP1 supports · met
Why
Variant confirmed in affected family cohort; segregation data support PP1 at supporting level despite incomplete penetrance noted in follow-up study.
Val412Ile is a heterozygous transition in exon 11 (g.69148G>A according to GenBank accession number AF109907.1; c.1234 according to GenBank accession number NM 000021.2) of the PSEN1 gene in the first position of codon 412, which predicts an aminoacidic substitution from valine to isoleucine and it was detected in a 38-year-old female (FUS family).
Location Abstract; Results, sections 3.1-3.2; Figure 2; Figure 3A pedigree; Discussion · Context DHPLC screening and Sanger sequencing of PSEN1 exons 3-12 in 17 f-EOFTD patients; RFLP and DHPLC for control validation · full text
PSEN1 and PRNP gene mutations: co-occurrence makes onset very early in a family with FTD phenotype.
Searched
c.1234G>Ap.Val412IleV412IVal412Ilec.1234
Found
Follow-up study of the FUS family originally reported in Bernardi et al. 2009. Identified a novel seven extra-repeat PRNP insertional mutation co-occurring with PSEN1 Val412Ile. PSEN1 Val412Ile alone was found in 2 affected and 2 cognitively healthy carriers (ages 49 and 71), demonstrating incomplete penetrance. Double carriers (PSEN1 + PRNP) had very early onset within the third decade. Both mutations showed variable penetrance when present separately.
Variant
✓ Names this variant — characterised directly
Applied to
→PP1 supports · met
Why
Documented incomplete penetrance (2 healthy carriers at ages 49 and 71) which tempers PP1 segregation evidence. Co-occurring PRNP mutation does not explain all affected cases, so BP5 not met.
The PSEN1 Val412Ile mutation, which was previously detected in the three patients [14], was also identified in two additional family members (IV-5, III-2, in this paper).
Location Abstract; Results; Table 1; Figure 1 pedigree; Discussion · Context Bidirectional sequencing of PRNP and PSEN1 genes; APOE genotyping, MAPT haplotype analysis, PRNP M129V polymorphism assessment · full text
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
27930341 ↗
Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase.
CLINVAR
29091718 ↗
Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer Disease.
CLINVAR