Classification rationale
BA1BS1BP4BP6
Benign
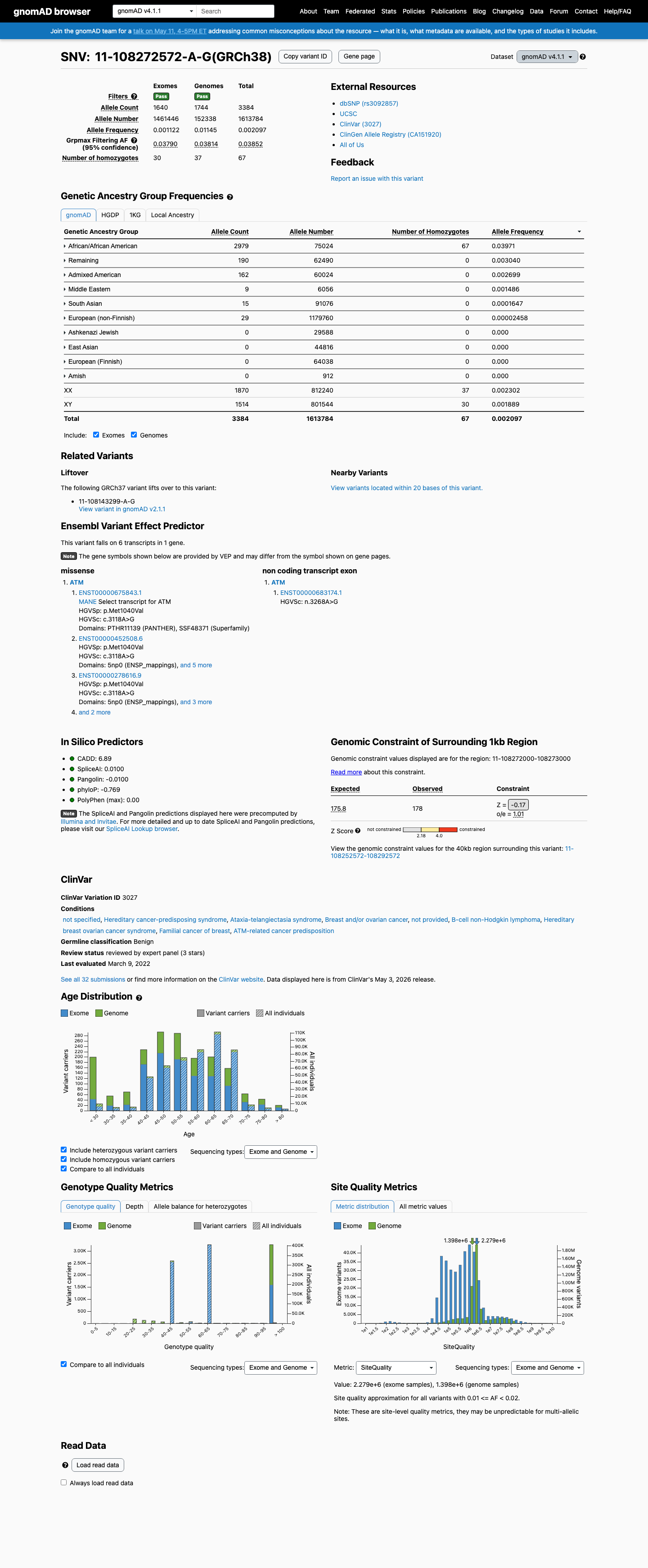
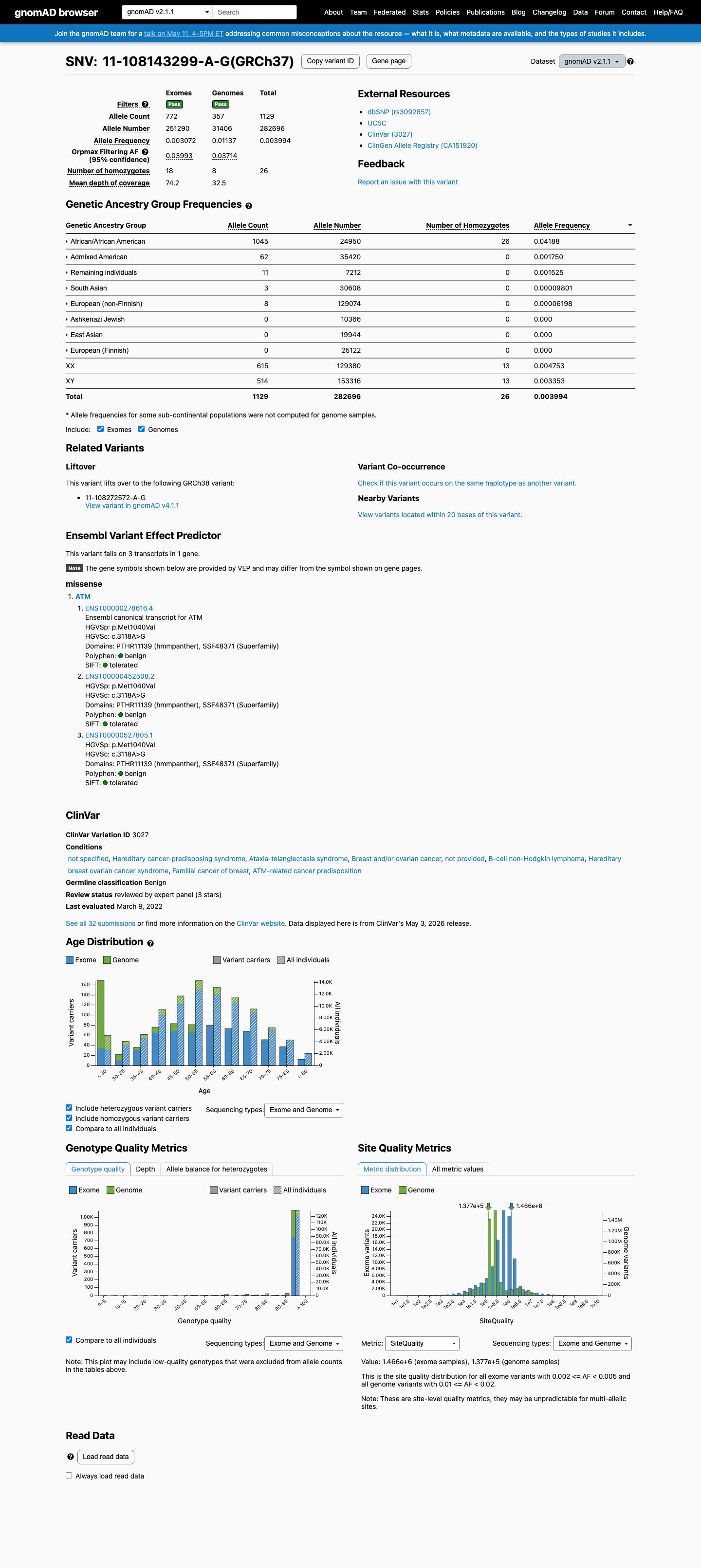
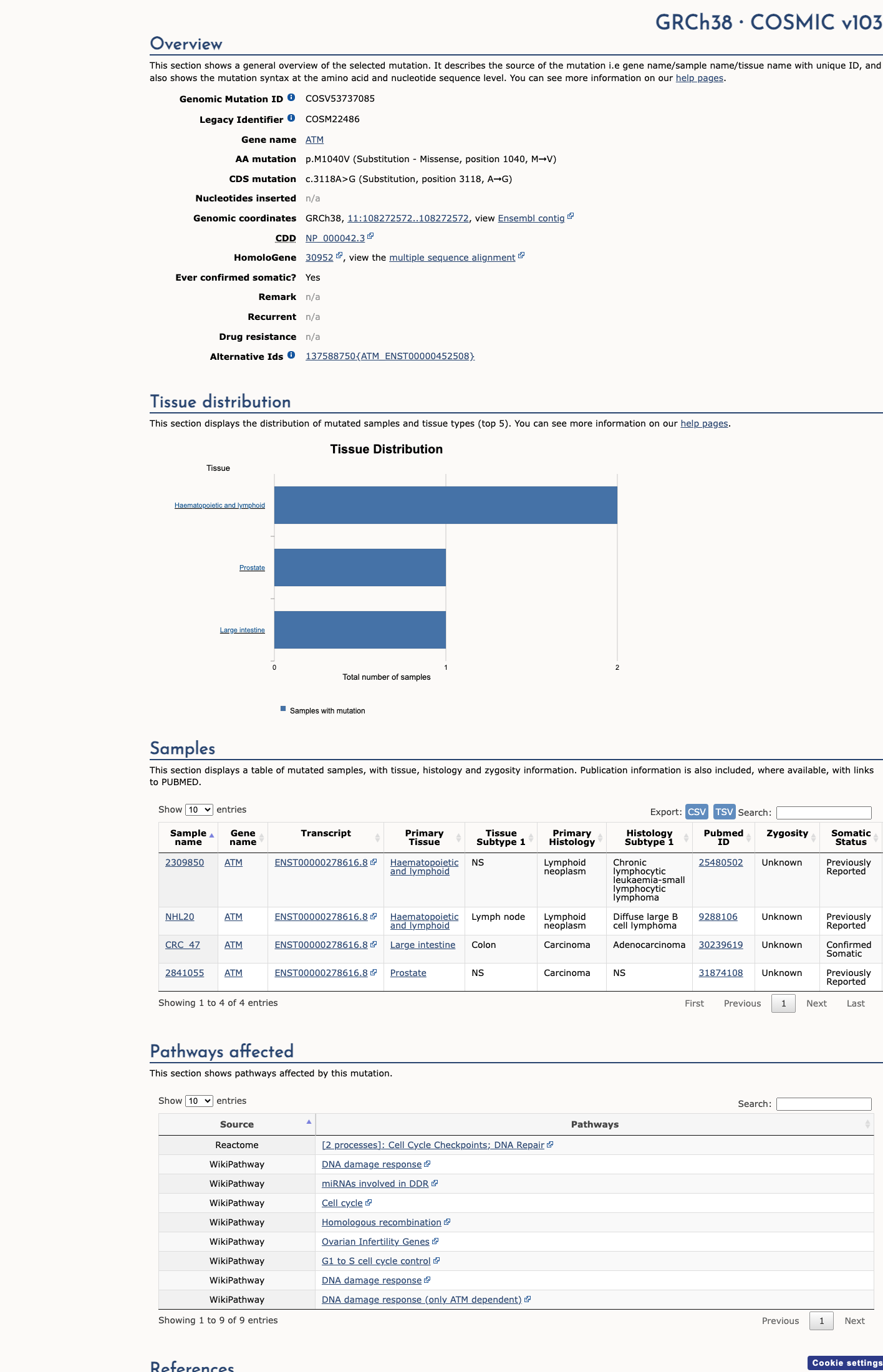
ATM c.3118A>G
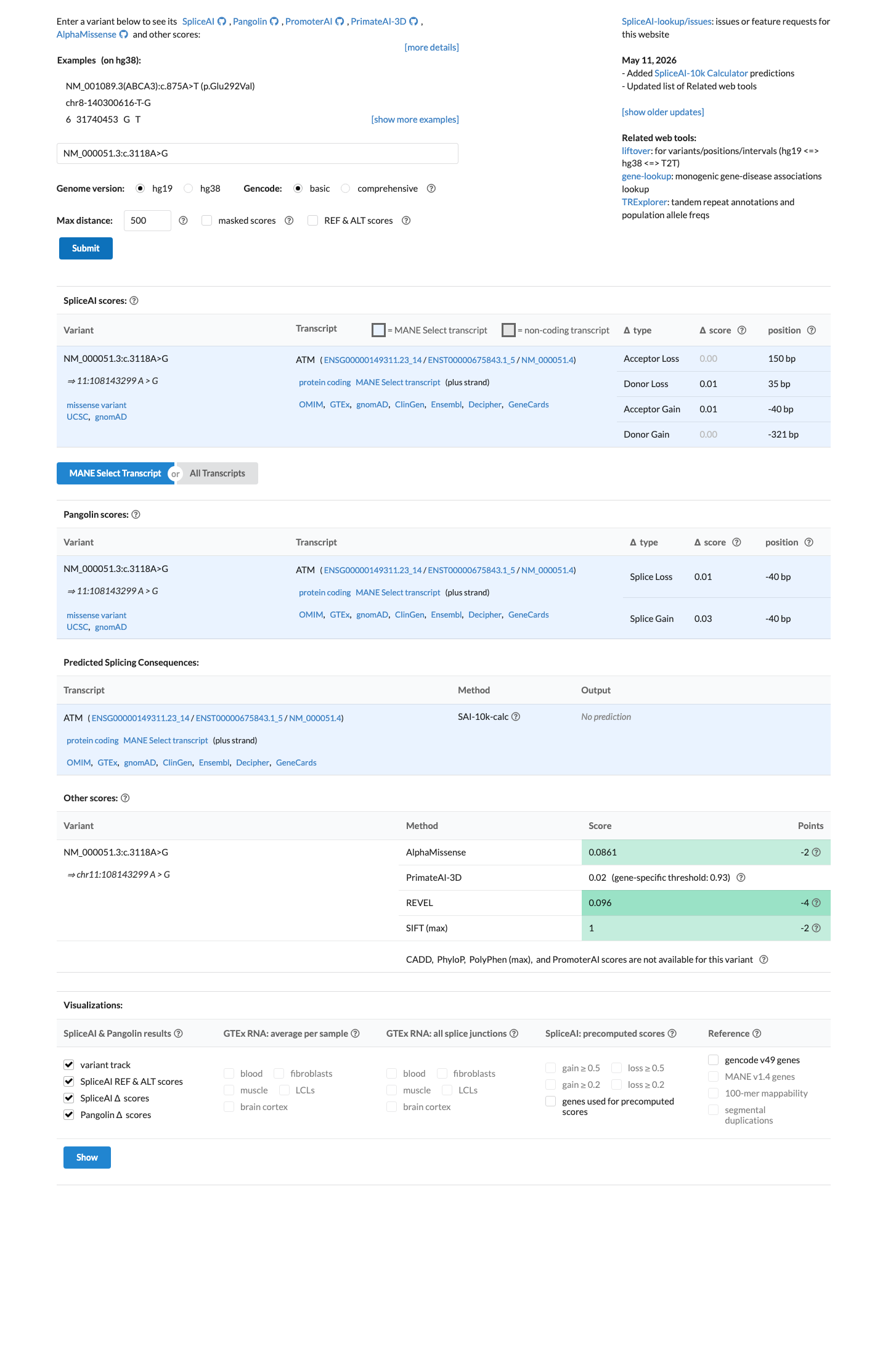
The ATM c.3118A>G (p.Met1040Val) variant has been observed in somatic cancers 4 times in COSMIC and is reported in ClinVar as Benign with expert panel review.1 This variant is common in population databases, with gnomAD v4.1 showing an overall allele frequency of 0.20969% and an African/African American allele frequency of 3.97073%, which exceeds the ATM BA1 threshold of 0.5% grpmax filtering allele frequency.2 Computational evidence supports a benign effect, with REVEL 0.096, BayesDel -0.451498, and SpliceAI maximum delta score 0.01, meeting the ATM BP4 thresholds and not meeting PP3 thresholds.3
BA1 + BS1 + BP4 + BP6
→
Benign