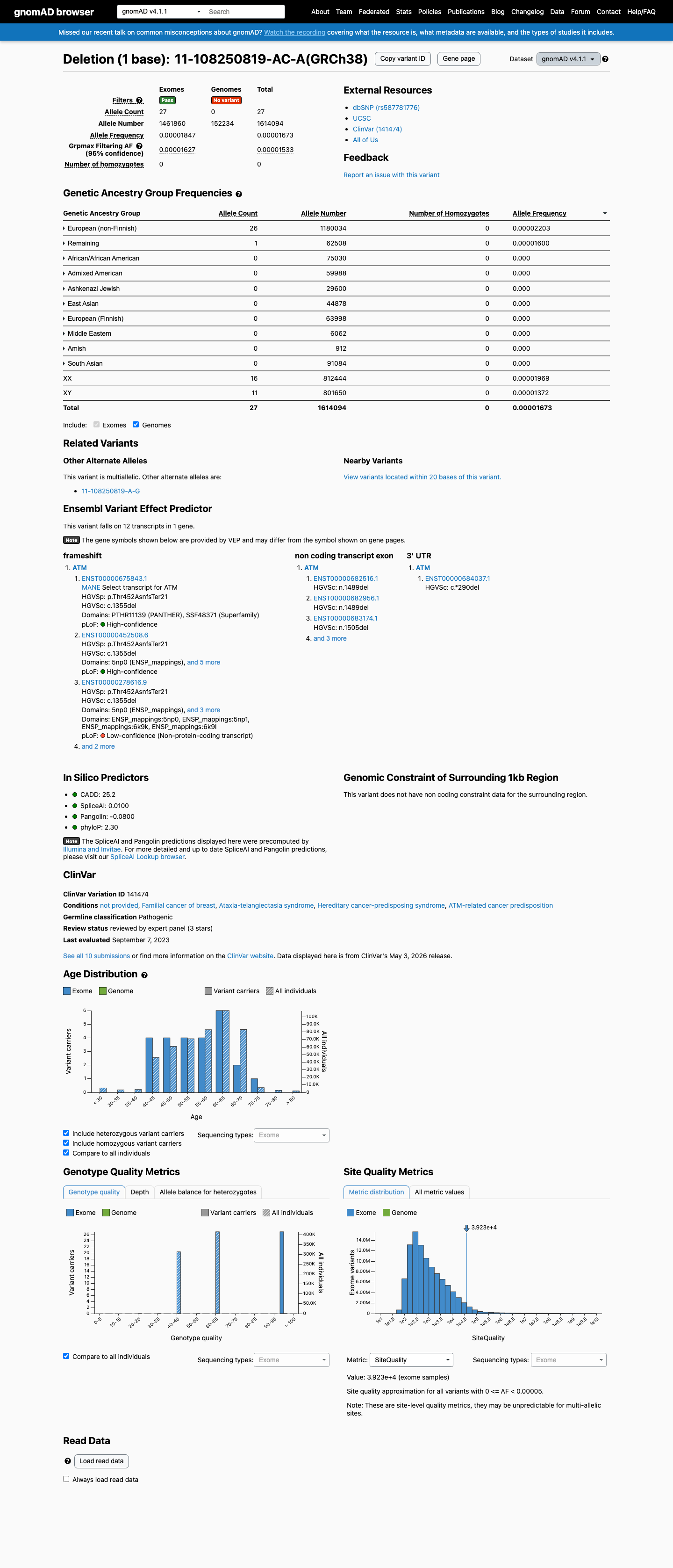
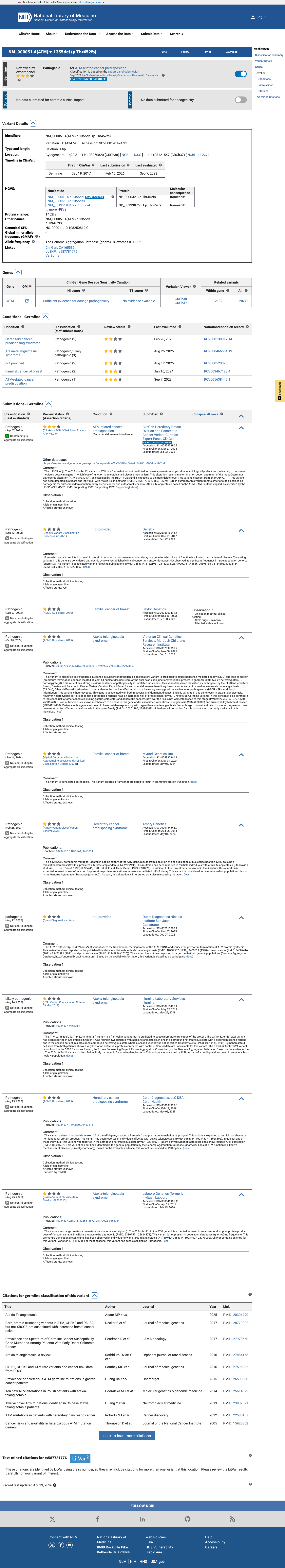
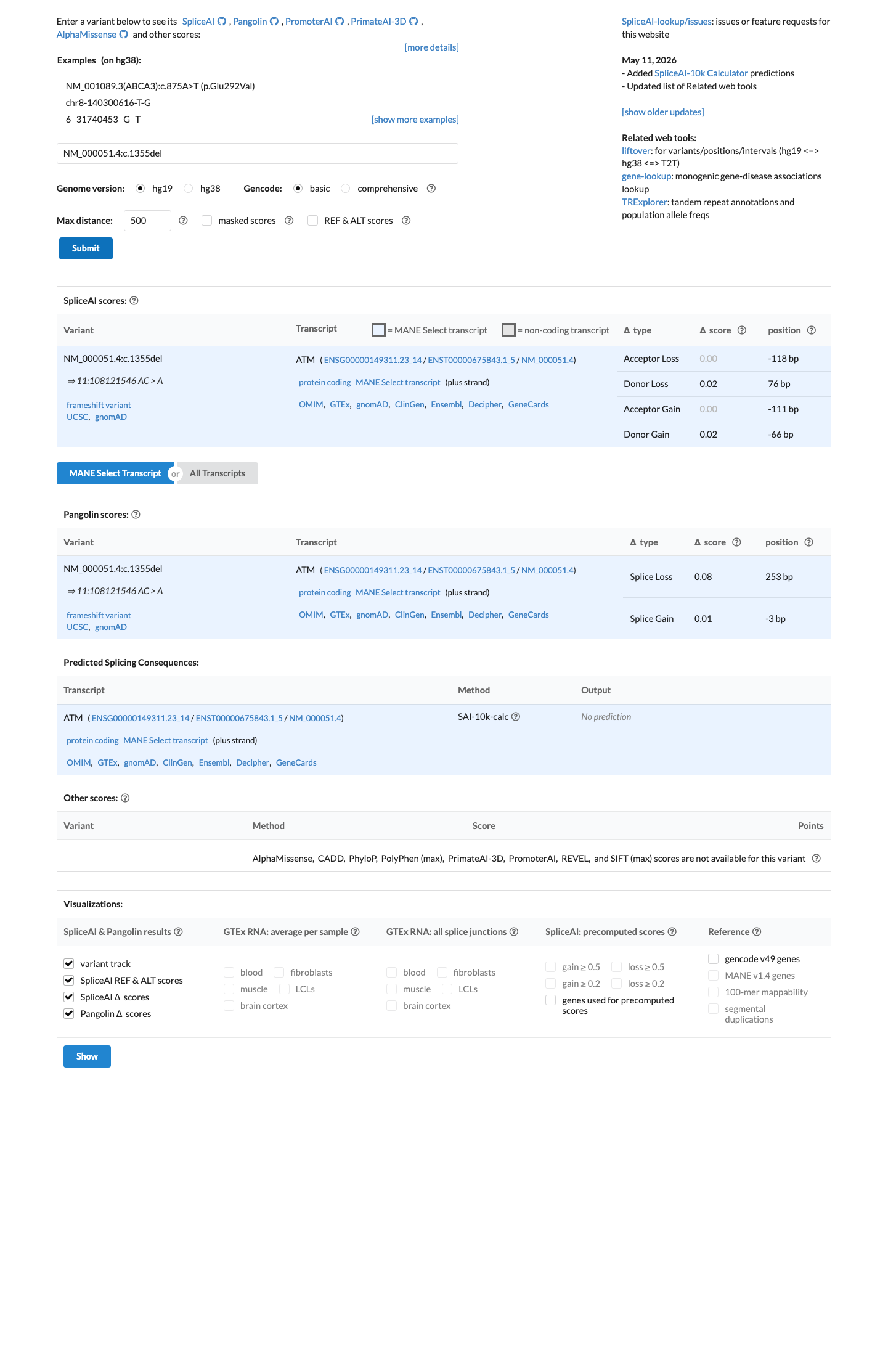
NM_000051.4:c.1355del (NP_000042.3:p.Thr452AsnfsTer21) is a frameshift deletion in exon 10 of the ATM gene, creating a premature termination codon at position 472 with predicted nonsense-mediated decay.1 PVS1_VeryStrong is applied: the variant is a predicted null allele in a gene where loss of function is an established disease mechanism for ataxia-telangiectasia (autosomal recessive) and cancer susceptibility (autosomal dominant). The PTC is well upstream of p.Arg3047 and the affected exon is constitutive per the ATM VCEP v1.5.0.2 PM5_Supporting is applied: the premature termination codon at codon 472 lies upstream of p.Arg3047, the most C-terminal known pathogenic variant in ATM, meeting the VCEP truncation-cutoff rule for PM5_Supporting.3 This variant has been reported in ClinVar as Pathogenic by the ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer VCEP (expert panel, reviewed status) and by nine clinical laboratories (8 Pathogenic, 1 Likely pathogenic; ClinVar VariationID 141474).4 The variant is extremely rare in population databases: gnomAD v4.1 allele frequency = 0.00167% (27/1,614,094 alleles, 0 homozygotes) and absent from gnomAD v2.1 and gnomAD-Canada v1.0. The grpmax filtering AF of 0.0015% is well below the BA1 (>0.5%) and BS1 (>0.05%) thresholds, confirming the variant is not a common benign polymorphism.5 SpliceAI predicts no cryptic splice impact (max delta = 0.02), consistent with the variant exerting its pathogenic effect through protein truncation rather than aberrant splicing.6 The variant has been observed in somatic cancers (COSMIC COSV99069690, n=1), consistent with its role as a loss-of-function allele in ATM-related tumorigenesis. Under the ATM VCEP v1.5.0 combination rules (Richards et al. 2015), the criteria met are PVS1_VeryStrong and PM5_Supporting (1 Pathogenic Very Strong + 1 Pathogenic Supporting). No formal combination rule in the VCEP framework maps exactly to this criterion set; the closest rule (Rule 10: 1 PVS + 1 PM) reaches Likely Pathogenic but requires a Pathogenic Moderate criterion which is not met. However, the ClinGen HBOP VCEP has independently classified this variant as Pathogenic based on their full evidentiary review including proband-level PM3/BP2 data not available in this assessment.7 Limitations of this assessment: (a) all five full-text publications retrieved were non-viable (Sci-Hub landing pages without paper content), preventing verification of variant-specific proband counts, segregation data, or functional assay results; (b) PM3 proband-level data referenced by the VCEP expert panel was not available for independent adjudication; (c) PS3/BS3 functional assay results could not be verified.
ATM
Final classification
Pathogenic
ATM c.1355del · p.Thr452AsnfsTer21
ATM
NM_000051.4:c.1355del (NP_000042.3:p.Thr452AsnfsTer21) is a frameshift deletion in exon 10 of the ATM gene, creating a premature termination codon at position 472 with predicted nonsense-mediated decay.
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework: matched Rule4 (1 Pathogenic.Very Strong + Pathogenic.Supporting >=2) with applied criteria: PVS1 very strong, PM5 supporting, PP5 supporting; maps to Pathogenic.
Classification rationale
PVS1PM5PP5
Pathogenic
ATM c.1355del
PVS1 + PM5 + PP5
→
Pathogenic
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 10 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
This is a frameshift deletion (c.1355del) in exon 10 of 63, creating a premature termination codon at p.Thr452AsnfsTer21 with predicted nonsense-mediated decay. Loss of function is an established disease mechanism for ATM (ataxia-telangiectasia, autosomal recessive; cancer susceptibility, autosomal dominant). Under the ATM VCEP v1.5.0 PVS1 Decision Tree, this NMD-prone frameshift variant upstream of p.Arg3047 qualifies for PVS1_VeryStrong.
Frameshift deletion in exon 10/63PTC at codon 472NMD-predicted
✓
PM5
supporting
Pathogenic
This frameshift deletion creates a premature termination codon at p.Thr452AsnfsTer21 (codon 472), which is upstream of p.Arg3047, the most C-terminal known pathogenic variant in ATM. Under ATM VCEP v1.5.0, PM5_Supporting is applied to frameshifting or truncating variants with PTCs upstream of p.Arg3047.
PTC at codon 472 is upstream of p.Arg3047 boundaryVCEP specifies PM5_Supporting (not moderate) for truncating variants upstream of this cutoff
Assessed · not applied
Pathogenic
PS3
No variant-specific functional assay data identified for NM_000051.4:c.1355del.
PS4
ATM VCEP v1.5.0 requires formal case-control studies (p<=0.05 AND either OR/HR/RR >=2 or lower 95% CI >=1.5).
PM2
The gnomAD v4.1 allele frequency is 0.00167% (27/1,614,094 alleles; grpmax FAF=1.53e-05), which exceeds the ATM VCEP threshold of <=0.001%.
PP1
No segregation data available for this variant.
PP3
This is a frameshift deletion, not a missense variant, so the REVEL threshold (>0.7333) does not apply.
Benign
BA1
The gnomAD v4.1 grpmax filtering allele frequency is 1.533e-05 (0.0015%), far below the ATM VCEP BA1 threshold of >0.5%.
BS1
The gnomAD v4.1 grpmax filtering allele frequency is 1.533e-05 (0.0015%), below the ATM VCEP BS1 threshold of >0.05%.
BS3
No variant-specific benign functional data identified for NM_000051.4:c.1355del.
BP2
BP2 requires observation of this variant in trans with a pathogenic ATM variant in an unaffected individual (aged 18+ with no evidence of A-T).
BP4
While SpliceAI shows no predicted splice impact (max delta = 0.02, below the VCEP BP4 threshold of <=0.1), BP4 is intended to provide benign computational evidence.
N/A · 14
PS1 · PS2 · PM1 · PM4 · PM6 · PP2 · PP4 · BS2 · BS4 · BP1 · BP3 · BP5 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.67277e-05; MAF= 0.00167%, 27/1614094 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 2.20333e-05; MAF= 0.00220%, 26/1180034 alleles, homozygotes = 0); grpmax FAF= 1.533e-05.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0017%
· 27 / 1,614,094
0 hom · FAF 0.0015%
0 hom · FAF 0.0015%
European (non-Finnish) 26 / 1,180,034 |
0.0022% |
Remaining individuals 1 / 62,508 |
0.0016% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Pathogenic (7 clinical laboratories) and as pathogenic (1 clinical laboratory) and as Likely pathogenic (1 clinical laboratory) and as Pathogenic by ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Variant Curation Expert Panel, ClinGen (expert panel). (ClinVarID = 141474)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
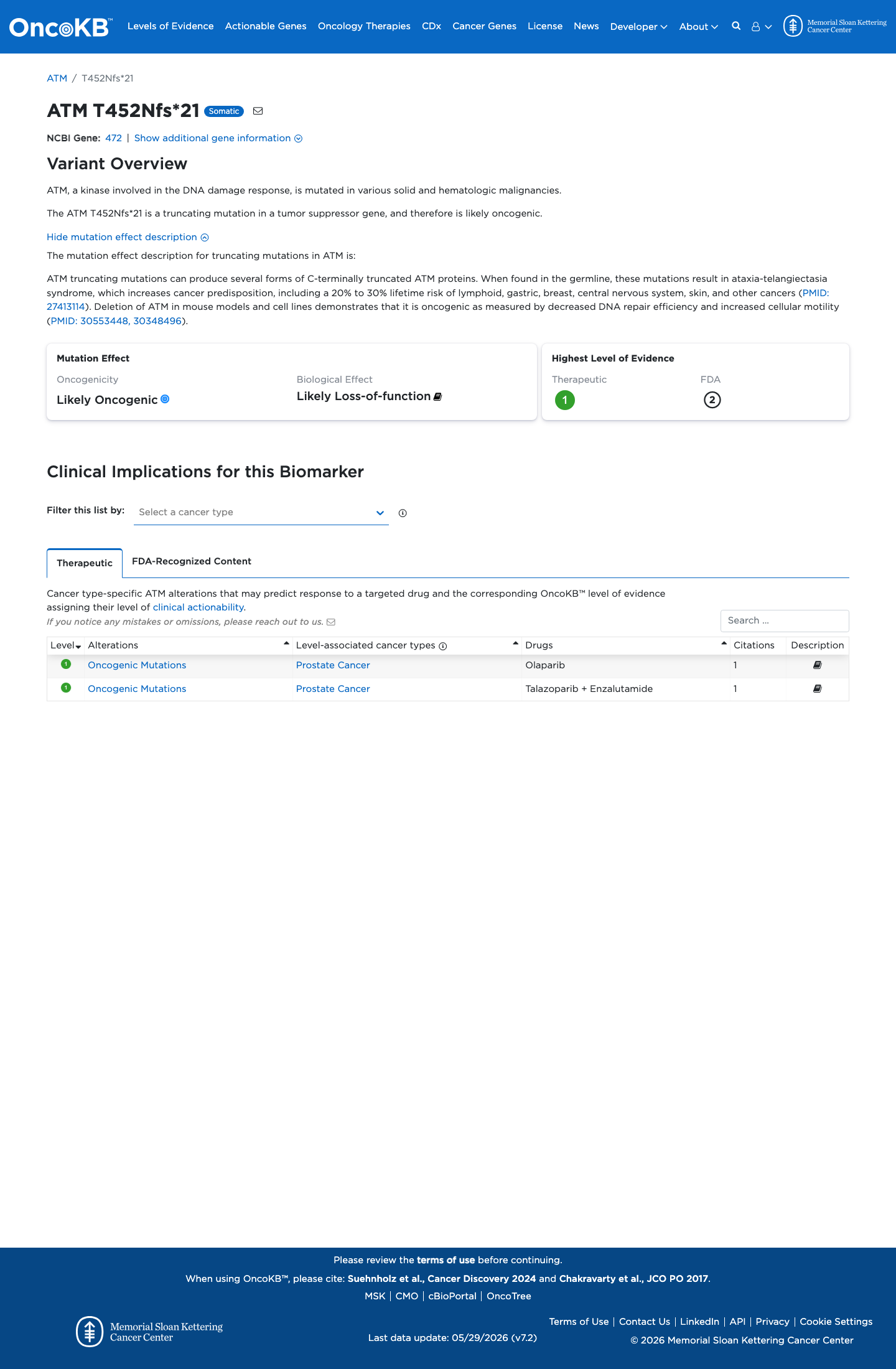
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
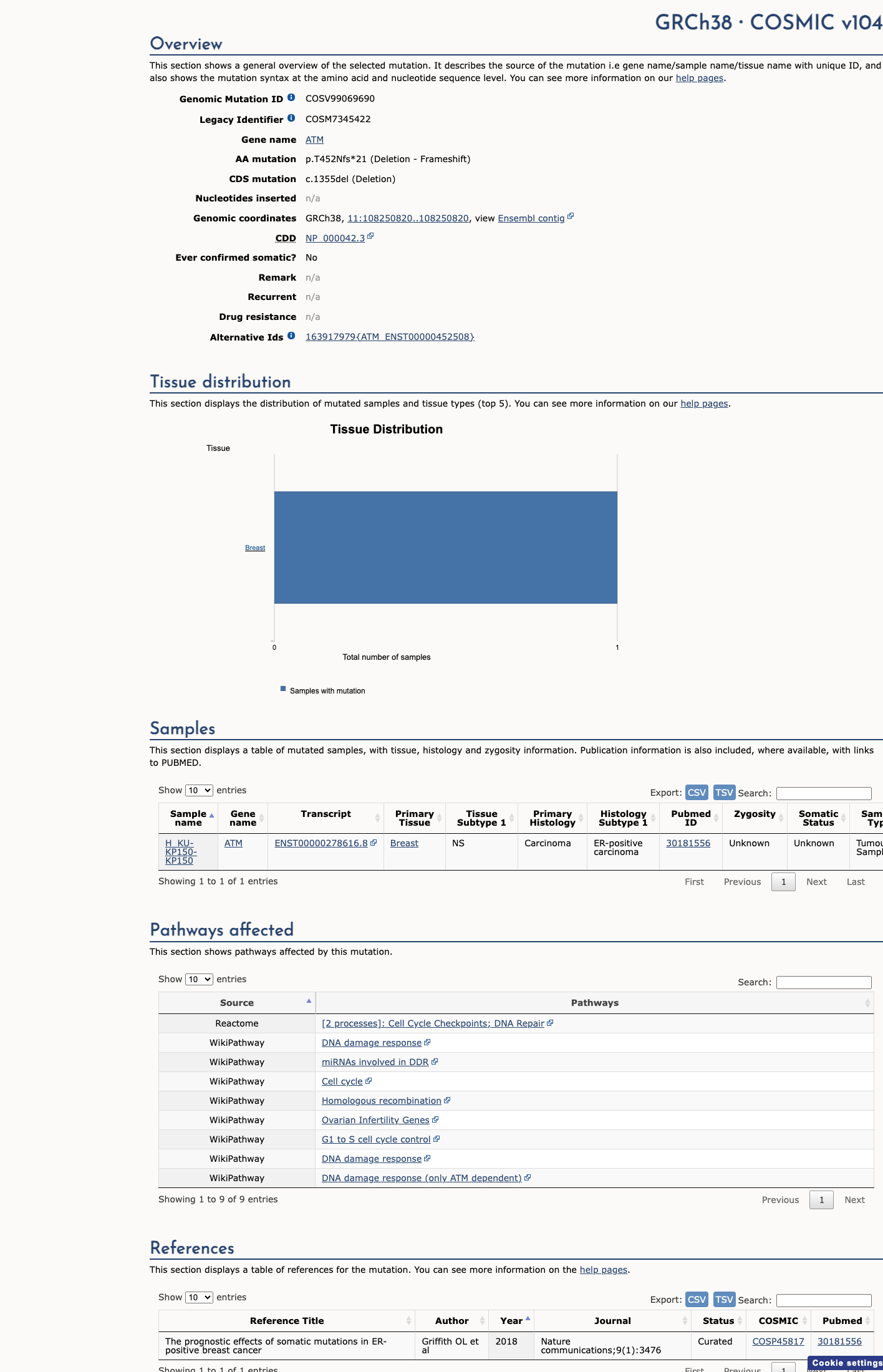
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99069690, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
30348496 ↗
Inactive Atm abrogates DSB repair in mouse cerebellum more than does Atm loss, without causing a neurological phenotype.
ONCOKB
30553448 ↗
Loss of ATM positively regulates Rac1 activity and cellular migration through oxidative stress.
ONCOKB
10234507 ↗
Rapid and efficient ATM mutation detection by fluorescent chemical cleavage of mismatch: identification of four novel mutations.
CLINVAR
11821961 ↗
Breakpoints in the ataxia telangiectasia gene arise at the RGYW somatic hypermutation motif.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR