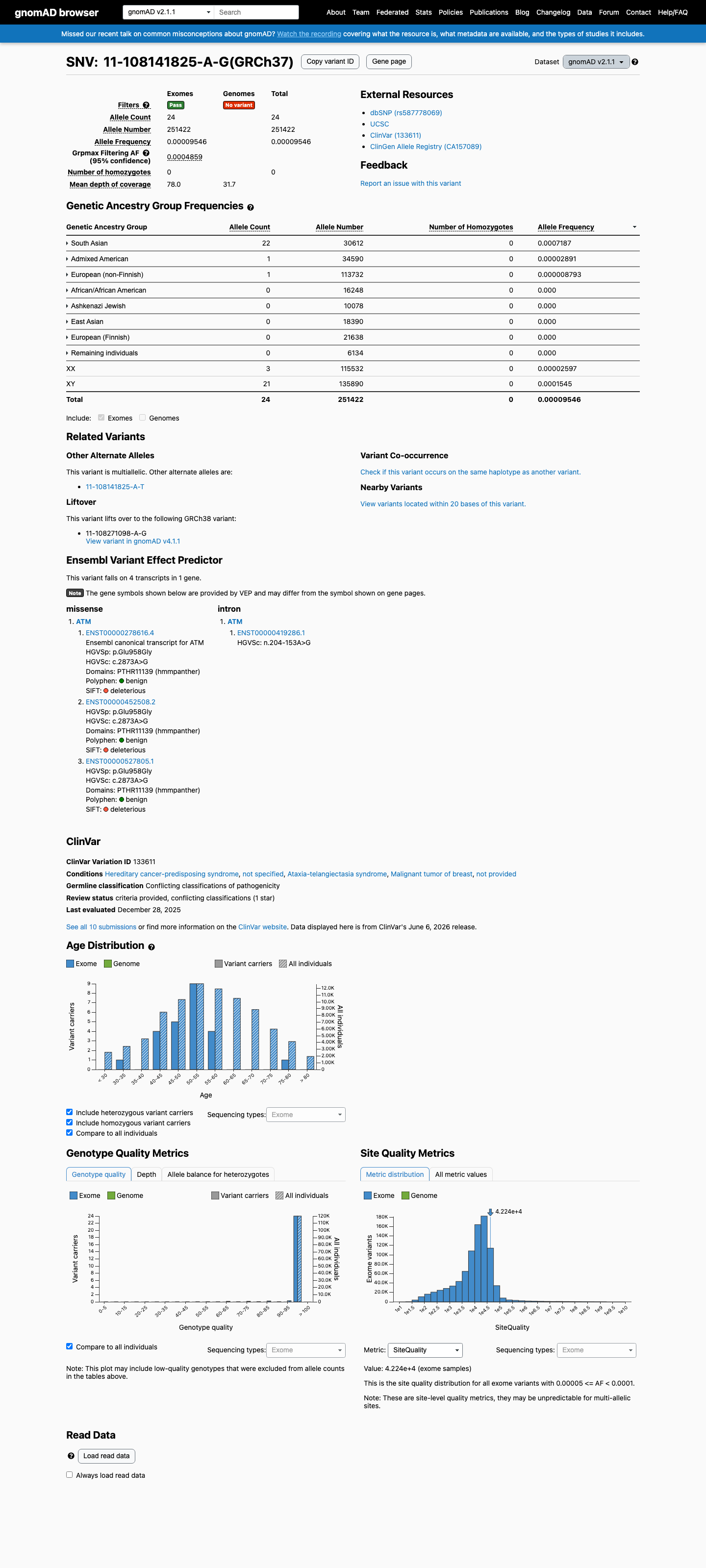
BS1_Strong: The variant is present in gnomAD v4.1 at a grpmax filtering allele frequency of 0.0752%, exceeding the ATM VCEP threshold of >0.05% for BS1. The variant has been observed on 90 alleles including one homozygous individual, with highest frequency in the South Asian population (0.091%).1 BS3_Moderate: Comprehensive functional assessment of all ATM SNVs using prime editing and deep learning (PMID:40580951) classifies p.Glu958Gly as 'Functional' with high confidence. The variant rescues both ATM-specific phosphorylation activity and radiosensitivity, meeting VCEP criteria for BS3_Moderate.2 BP2_Strong: c.2873A>G has been observed in cis with a known pathogenic ATM variant (c.5932G>T, p.Glu1978*) on the same allele, corresponding to -4.0 points (BP2_Strong) under the ATM VCEP PM3/BP2 table.3 BP4_Supporting: REVEL score of 0.121 is below the VCEP benign threshold of ≤0.249. BayesDel score of -0.224 is consistent with a benign prediction. SpliceAI predicts no significant splice impact (max delta 0.19).4 No pathogenic criteria were met: PVS1 is not applicable (missense variant). PS1 is not met (no same amino acid change classified as pathogenic at codon 958). PS3 is not met (functional studies show normal, not damaging, effect). PM2 is not met (AF=0.00558% exceeds ≤0.001% VCEP threshold). PP3 is not met (REVEL=0.121 below >0.7333 threshold; SpliceAI=0.19 below ≥0.2 threshold).5
ATM
Final classification
Benign
ATM c.2873A>G · p.Glu958Gly
ATM
BS1_Strong: The variant is present in gnomAD v4.1 at a grpmax filtering allele frequency of 0.0752%, exceeding the ATM VCEP threshold of >0.05% for BS1. The variant has been observed on 90 alleles including one homozygous individual, with highest frequency in the South Asian population (0.091%).
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework: matched Rule16 (Benign.Strong >=2) with applied criteria: BS1 strong, BS3 moderate, BP2 strong, BP4 supporting; maps to Benign.
Classification rationale
BS1BS3BP2BP4
Benign
ATM c.2873A>G
BS1 + BS3 + BP2 + BP4
→
Benign
2
PMID:40580951 ↗vcep_suppl_tables1_pmid_40580951
3
vcep_atm_pm3_bp2_1_5
4
revelbayesdelspliceai ↗
5
revelspliceai ↗gnomad_v4 ↗vcep_suppl_tables1_pmid_40580951
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 7 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
gnomAD v4.1 grpmax filtering allele frequency is 0.0752% (FAF=0.000752), exceeding the VCEP BS1 threshold of >0.05%. The variant is present on 90 alleles including one homozygous individual, indicating it is more common than expected for a highly penetrant ATM pathogenic variant.
gnomAD v4.1: grpmax FAF=0.000752 (0.0752%)exceeding VCEP BS1 threshold of >0.05%. 90 alleles1 homozygote. Highest in South Asian population (AF=0.0911%).
✓
BS3
moderate
Benign
Comprehensive functional assessment of all ATM SNVs using prime editing and deep learning (PMID:40580951) classifies c.2873A>G (p.Glu958Gly) as 'Functional' with high confidence (combined score -0.866). The variant rescues both ATM-specific phosphorylation activity and radiosensitivity, meeting VCEP BS3_Moderate criteria. Prior independent studies (Barone et al. 2009, Mitui et al. 2009) also reported normal kinase activity and radiosensitivity for this variant.
PMID:40580951 Suppl Table S1: E958G classified as 'Functional'High confidencecombined score -0.866. Rescues both ATM-specific phosphorylation and radiosensitivity. Prior studies corroborate normal function.
✓
BP2
strong
review
Benign
c.2873A>G has been observed in cis with a known pathogenic ATM variant (c.5932G>T, p.Glu1978*) on the same allele. Under the ATM VCEP PM3/BP2 table, confirmed cis configuration with a pathogenic variant in a recessive gene assigns -4.0 points, corresponding to BP2_Strong.
Observed in cis with pathogenic ATM variant c.5932G>T (p.Glu1978*). VCEP BP2 table: confirmed in cis = -4.0 points = BP2_Strong.
✓
BP4
supporting
Benign
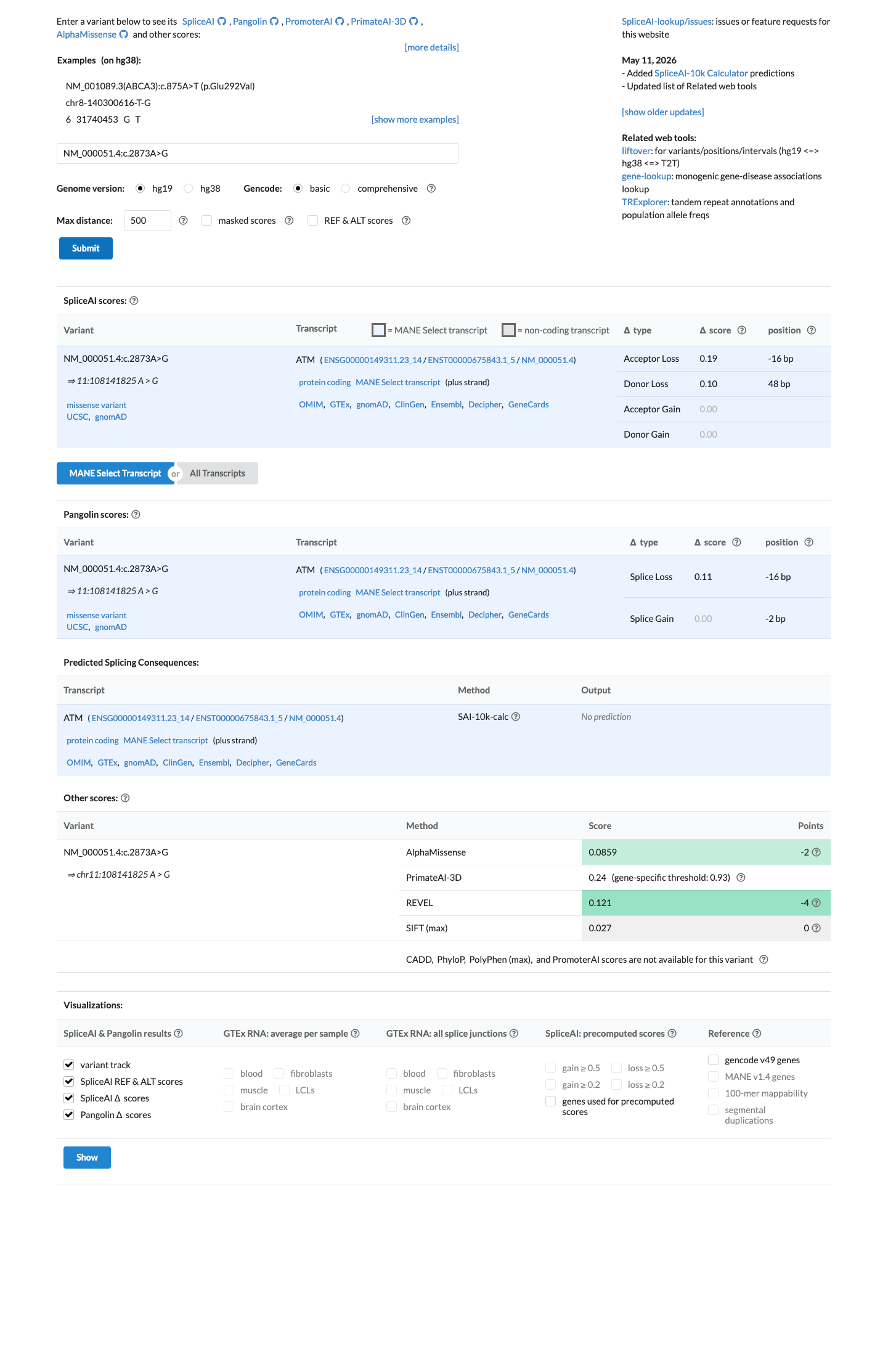
REVEL score is 0.121, below the VCEP BP4 missense threshold of ≤0.249. BayesDel score is -0.224, consistent with a benign computational prediction. SpliceAI predicts no significant splice impact (max delta 0.19).
REVEL=0.121 (≤0.249 VCEP BP4 threshold). BayesDel=-0.224 (benign). SpliceAI max_delta=0.19 (no significant splice impact).
Assessed · not applied
Pathogenic
PS1
No known pathogenic or likely pathogenic variant with the same amino acid change (p.Glu958Gly) has been identified.
PS3
Well-established functional studies demonstrate normal ATM kinase activity and normal radiosensitivity for p.Glu958Gly, supporting a benign functional effect rather than a damaging one.
PS4
No case-control study or large case series demonstrating statistically significant enrichment of c.2873A>G in affected individuals compared to controls has been identified.
PM2
gnomAD v4.1 allele frequency is 5.58×10⁻⁵ (0.00558%, 90/1,614,012 alleles), which exceeds the VCEP PM2_Supporting threshold of ≤0.001% (≤1×10⁻⁵).
PP1
No published co-segregation data for c.2873A>G in families with multiple affected members has been identified.
PP3
REVEL score is 0.121, well below the VCEP PP3 threshold of >0.7333 for missense variants.
Benign
BA1
gnomAD v4.1 grpmax filtering allele frequency is 0.0752% (FAF=0.000752), which does not exceed the VCEP BA1 threshold of >0.5%.
N/A · 17
PVS1 · PS2 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP4 · PP5 · BS2 · BS4 · BP1 · BP3 · BP5 · BP6 · BP7
Research & evidence
Population frequency · supports benign

gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 5.57617e-05; MAF= 0.00558%, 90/1614012 alleles, homozygotes = 1) and has highest observed frequency in the South Asian population (AF= 0.000911207; MAF= 0.09112%, 83/91088 alleles, homozygotes = 1); grpmax FAF= 0.00075231.
v2.1
This variant is present in gnomAD v2.1 (AF= 9.5457e-05; MAF= 0.00955%, 24/251422 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.000718672; MAF= 0.07187%, 22/30612 alleles, homozygotes = 0); grpmax FAF= 0.00048585.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0056%
· 90 / 1,614,012
1 hom · FAF 0.075%
1 hom · FAF 0.075%
South Asian 83 / 91,088 |
0.091% 1 hom |
Remaining individuals 3 / 62,488 |
0.0048% |
Admixed American 1 / 59,996 |
0.0017% |
African/African American 1 / 74,918 |
0.0013% |
European (non-Finnish) 2 / 1,180,036 |
0.00017% |
+ 5 not observed (European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0095%
· 24 / 251,422
0 hom · FAF 0.049%
0 hom · FAF 0.049%
South Asian 22 / 30,612 |
0.072% |
Admixed American 1 / 34,590 |
0.0029% |
European (non-Finnish) 1 / 113,732 |
0.00088% |
+ 5 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
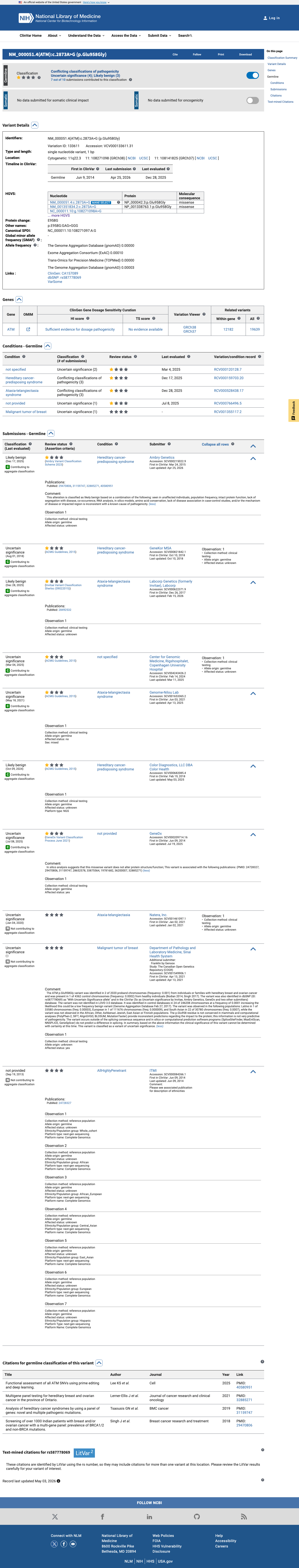
This variant has been reported in ClinVar as Uncertain significance (6 clinical laboratories) and as Likely benign (3 clinical laboratories). (ClinVarID = 133611)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.19). REVEL score = 0.121. BayesDel score = -0.224243.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. ATM, a kinase involved in the DNA damage response, is mutated in various solid and hematologic malignancies.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 9 further PMIDs triaged but not cited — see Sources & References.
Functional assessment of all ATM SNVs using prime editing and deep learning.
Found
Suppl Table S1: E958G classified as 'Functional' High confidence combined score -0.866.
Applied to
→BS3 supports · met
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
24418350 ↗
EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood.
CLINVAR
24728327 ↗
Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR