Classification rationale
PP3
Likely Pathogenic
ATM c.3137T>C
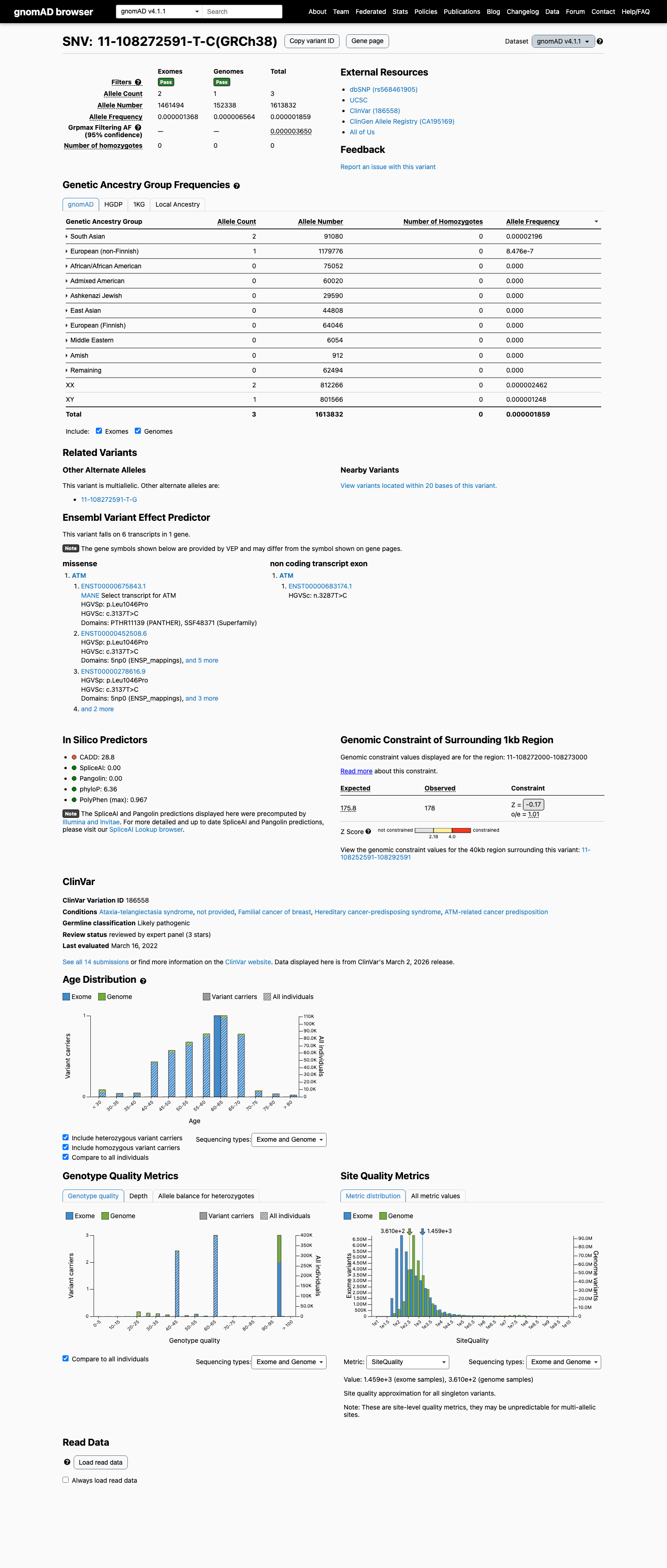
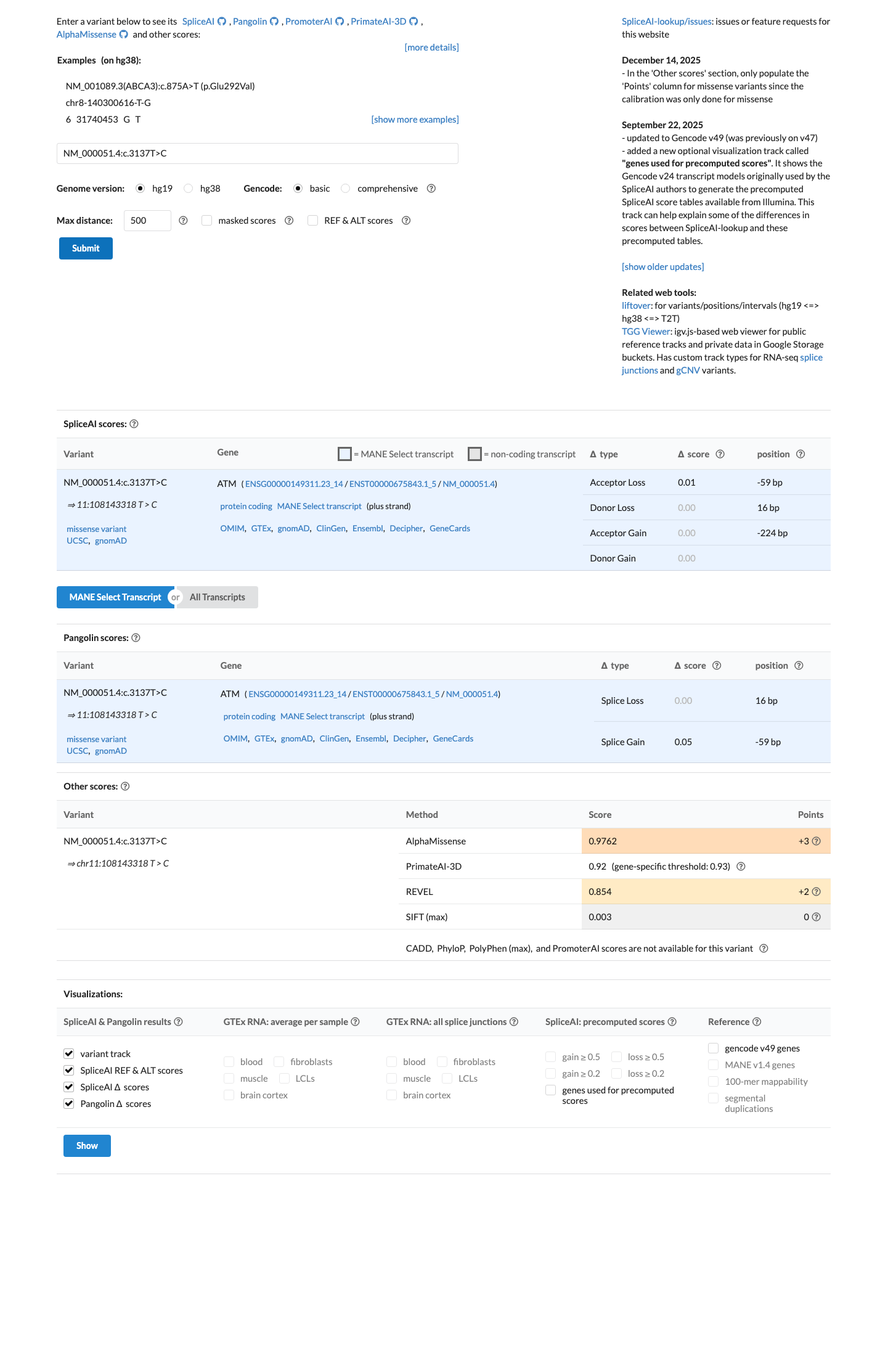
The ATM c.3137T>C (p.Leu1046Pro) variant has not been observed in somatic cancers in COSMIC and has not been reported in ClinVar.1 This variant is absent from gnomAD v2.1 and is present at very low frequency in gnomAD v4.1 overall (AF 1.85893e-06; 3/1613832 alleles; 0 homozygotes), with the highest observed frequency in the South Asian population at 2.19587e-05 (2/91080 alleles; 0 homozygotes).2 In silico evidence supports a deleterious missense effect, with REVEL 0.854 exceeding the ATM PP3 threshold of 0.7333, while SpliceAI predicts no significant splice impact (maximum delta score 0.01).3
PP3
→
Likely Pathogenic
1
evidence.json:cosmicevidence.json:clinvar
2
evidence.json:gnomad
3
prefetch.json:revelevidence.json:spliceaicase_summary.json:cspec