NM_000051.4:c.7357C>T (p.Arg2453Cys) is a missense variant in ATM exon 50, observed at very low frequency in population databases (gnomAD v4.1: 26/1,614,000 alleles, AF = 0.00161%; gnomAD v2.1: 3/251,250 alleles, AF = 0.00119%).1 The variant meets PM2_supporting per the ClinGen HBOP VCEP v1.5.0 fallback rule: although global gnomAD v4 frequency (0.00161%) exceeds the primary threshold of ≤0.001%, the variant is observed at n=1 in the East Asian subpopulation, which is sufficiently rare to warrant supporting-level evidence for pathogenicity.2 No pathogenic or likely pathogenic missense comparator at p.Arg2453 has been established (PS1 not met). REVEL score of 0.457 does not meet the VCEP PP3 threshold (>0.7333) or the BP4 threshold (≤0.249). SpliceAI predicts no splicing impact (max delta = 0.00).3 No variant-specific functional assay data (PS3/BS3) or segregation data (PP1) are available. The VCEP functional assay reference tables do not include this variant. The VCEP Suppl Table S1 (PMID 40580951) classifies the variant as 'Functional' with high confidence based on combined in silico scores, but computational predictions alone do not constitute experimental functional evidence sufficient for PS3.4 The variant has been reported in ClinVar as Uncertain Significance by 11 clinical laboratories (ClinVar Variation ID: 230366). No expert panel classification has been assigned. No peer-reviewed publication was identified that directly mentions NM_000051.4:c.7357C>T.5 The variant has been reported in COSMIC (COSV104592374, n=5) in somatic cancers, but this observation does not independently support germline pathogenicity classification under the HBOP VCEP framework. Applying the ClinGen HBOP VCEP v1.5.0 final classification rules (Richards et al. 2015 combining criteria), only PM2_supporting is met, with no benign criteria met. This does not reach the threshold for Likely Pathogenic (requires ≥2 supporting + ≥1 moderate, or ≥3 moderate, etc.) nor Likely Benign (requires ≥2 supporting benign). The variant is classified as Uncertain Significance.6
ATM
Final classification
VUS
ATM c.7357C>T · p.Arg2453Cys
ATM
NM_000051.4:c.7357C>T (p.Arg2453Cys) is a missense variant in ATM exon 50, observed at very low frequency in population databases (gnomAD v4.1: 26/1,614,000 alleles, AF = 0.00161%; gnomAD v2.1: 3/251,250 alleles, AF = 0.00119%).
Richards et.al., 2015 - Combining rules v1.5.0 criteria-combination framework was evaluated deterministically with applied criteria: PM2 supporting; no rule matched the adjudicated criteria.
Classification rationale
PM2
VUS
ATM c.7357C>T
PM2
→
VUS
3
revelspliceai ↗vcep_suppl_tables1_pmid_40580951
4
vcep_clingen_hbop_atm_supplementary_tables_1_and_2_v1vcep_suppl_tables1_pmid_40580951
6
cspec ↗generic_acmg_combination_rules
Gene diagram
· NM_000051.4 · variants mapped to exon structure
ATM
NM_000051.4
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 10 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
PM2_supporting applies under the VCEP fallback rule: although the global gnomAD v4.1 allele frequency (0.00161%, 26/1,614,000 alleles) exceeds the primary threshold of ≤0.001%, the variant is observed at n=1 in a single subpopulation (East Asian, AC=1, AN=44,878), which meets the VCEP criterion that a single-allele subpopulation occurrence is sufficiently rare to warrant PM2_supporting.
gnomAD v4.1: total AF = 0.00161% (26/1614000)
Assessed · not applied
Pathogenic
PS1
PS1 requires a known pathogenic missense variant at the same amino acid residue (p.Arg2453) with splicing ruled out for both.
PS3
No variant-specific experimental functional assay data were identified for NM_000051.4:c.7357C>T.
PS4
No case-control study data are available for NM_000051.4:c.7357C>T.
PP1
No segregation data are available for NM_000051.4:c.7357C>T.
PP3
The VCEP missense PP3 threshold requires REVEL >0.7333.
Benign
BA1
The VCEP BA1 threshold requires a grpmax filtering allele frequency >0.5% in gnomAD v4.
BS1
The VCEP BS1 threshold requires a grpmax filtering allele frequency >0.05% in gnomAD v4.
BS3
No variant-specific experimental functional assay data demonstrating a benign effect (rescue of ATM-specific features and/or radiosensitivity) were identified.
BP2
No evidence of co-occurrence in trans with a pathogenic or likely pathogenic ATM variant in an unaffected individual aged ≥18 years was identified.
BP4
The VCEP BP4 missense threshold requires REVEL ≤0.249.
N/A · 15
PVS1 · PS2 · PM1 · PM5 · PM6 · PP2 · PP4 · PP5 · BS2 · BS4 · BP1 · BP3 · BP5 · BP6 · BP7
Research & evidence
Population frequency
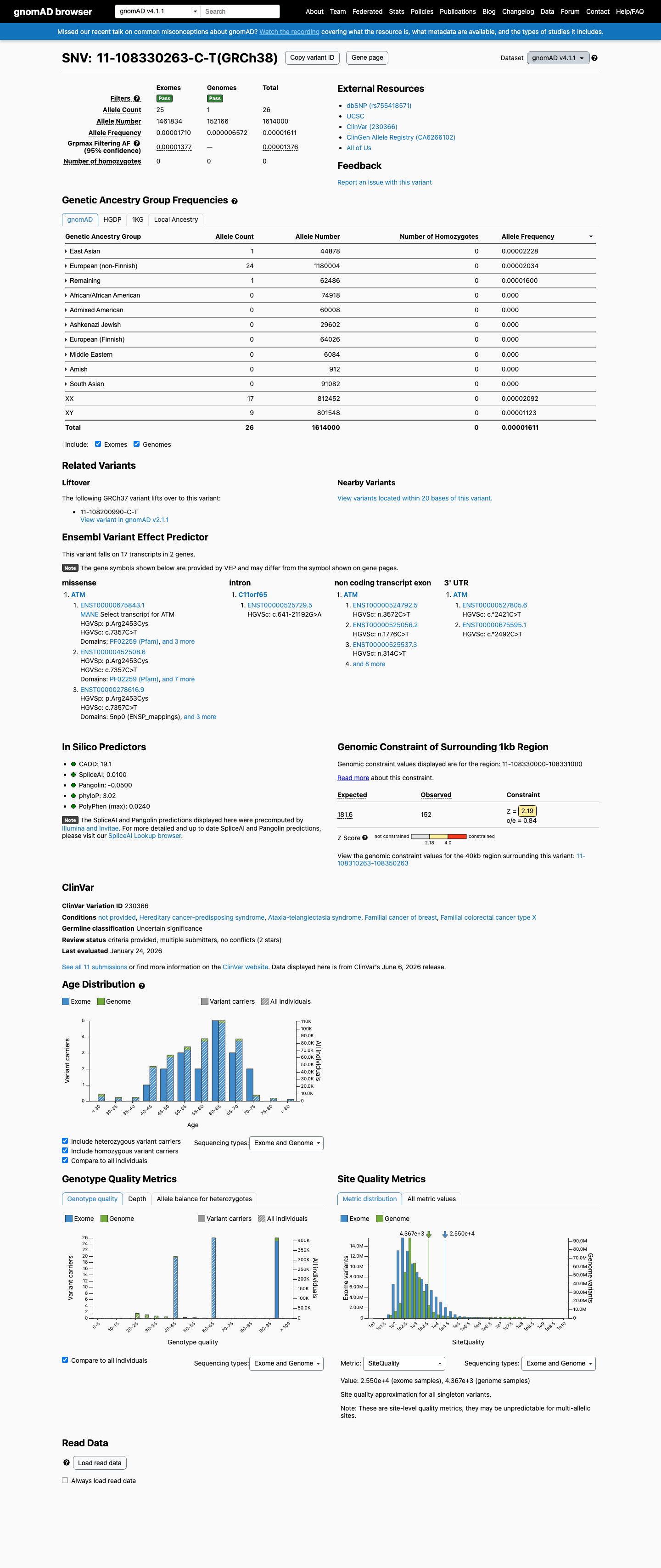
gnomAD v4.1
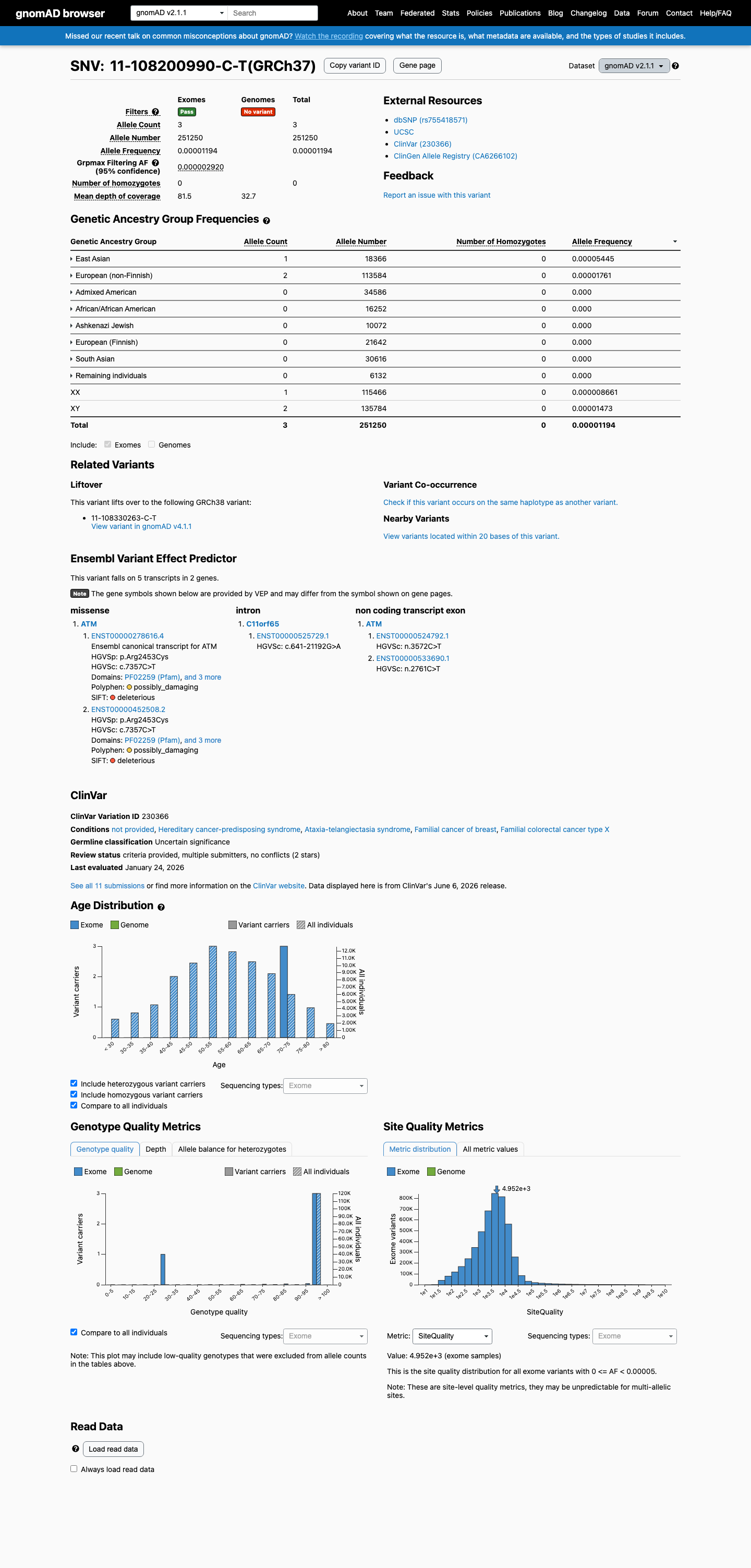
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.6109e-05; MAF= 0.00161%, 26/1614000 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 2.22826e-05; MAF= 0.00223%, 1/44878 alleles, homozygotes = 0); grpmax FAF= 1.376e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 1.19403e-05; MAF= 0.00119%, 3/251250 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 5.44484e-05; MAF= 0.00544%, 1/18366 alleles, homozygotes = 0); grpmax FAF= 2.92e-06.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0016%
· 26 / 1,614,000
0 hom · FAF 0.0014%
0 hom · FAF 0.0014%
East Asian 1 / 44,878 |
0.0022% |
European (non-Finnish) 24 / 1,180,004 |
0.002% |
Remaining individuals 1 / 62,486 |
0.0016% |
+ 7 not observed (Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0012%
· 3 / 251,250
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
East Asian 1 / 18,366 |
0.0054% |
European (non-Finnish) 2 / 113,584 |
0.0018% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
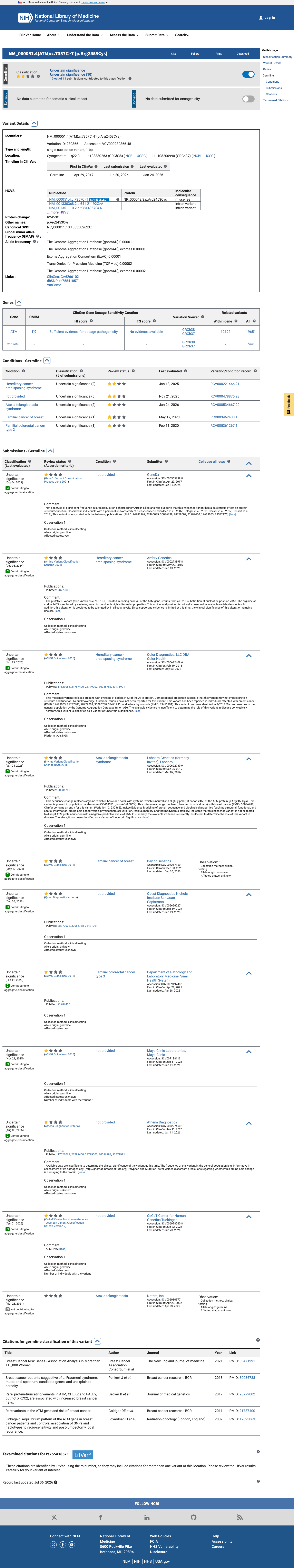
ClinVar
This variant has been reported in ClinVar as Uncertain significance (11 clinical laboratories). (ClinVarID = 230366)
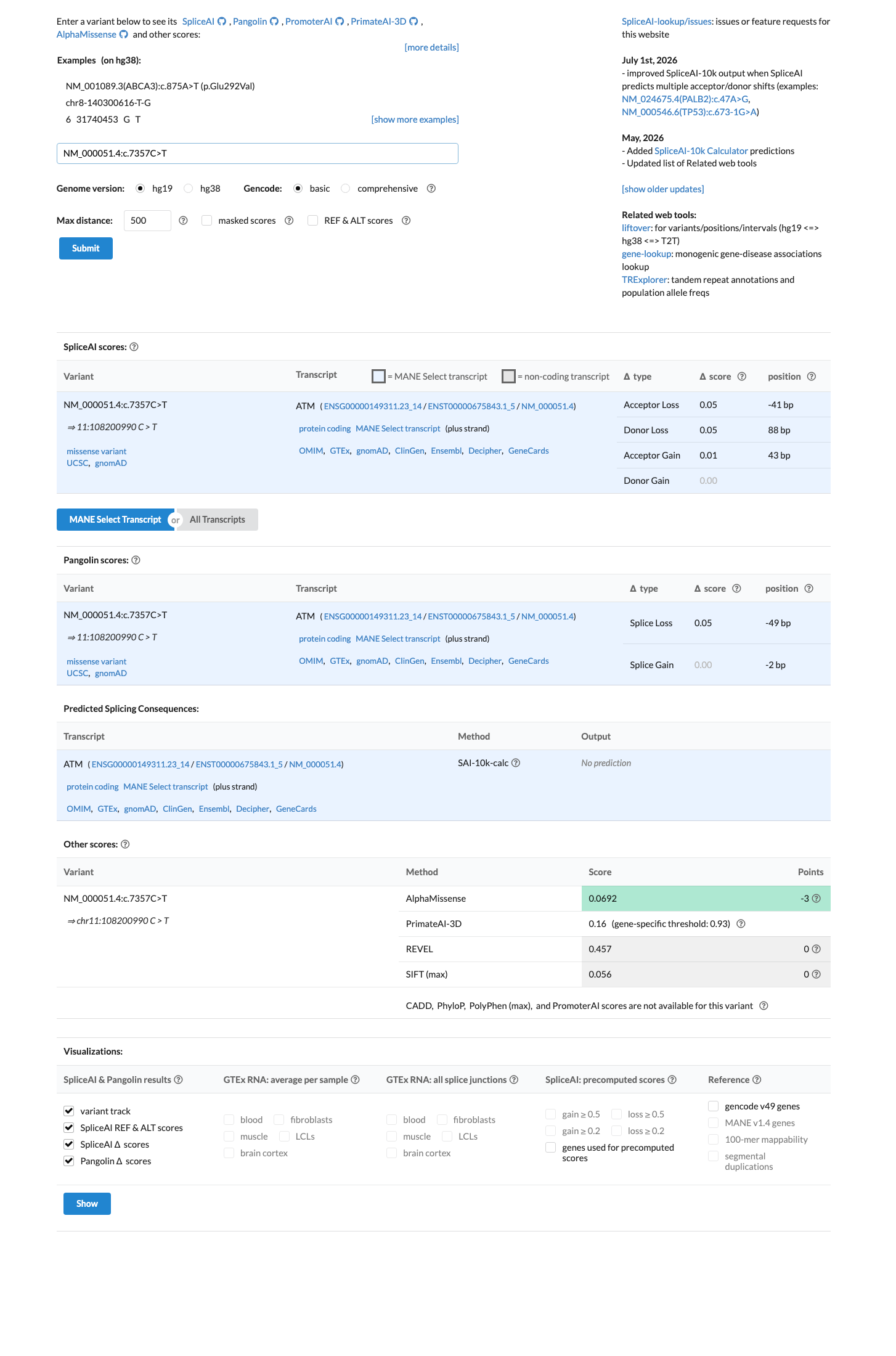
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.457. BayesDel score = -0.245392.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. ATM, a kinase involved in the DNA damage response, is mutated in various solid and hematologic malignancies.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV104592374, n = 5 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 8 PMIDs not cited in assessment
17623063 ↗
Linkage disequilibrium pattern of the ATM gene in breast cancer patients and controls; association of SNPs and haplotypes to radio-sensitivity and post-lumpectomy local recurrence.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
28779002 ↗
Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks.
CLINVAR